Axenfeld-Rieger Syndrome with Secondary Glaucoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Megalocornea Jeffrey Welder and Thomas a Oetting, MS, MD September 18, 2010
Megalocornea Jeffrey Welder and Thomas A Oetting, MS, MD September 18, 2010 Chief Complaint: Visual disturbance when changing positions. History of Present Illness: A 60-year-old man with a history of simple megalocornea presented to the Iowa City Veterans Administration Healthcare System eye clinic reporting visual disturbance while changing head position for several months. He noticed that his vision worsened with his head bent down. He previously had cataract surgery with an iris-sutured IOL due to the large size of his eye, which did not allow for placement of an anterior chamber intraocular lens (ACIOL) or scleral-fixated lens. Past Medical History: Megalocornea Medications: None Family History: No known history of megalocornea Social History: None contributory Ocular Exam: • Visual Acuity (with correction): • OD 20/100 (cause unknown) • OS 20/20 (with upright head position) • IOP: 18mmHg OD, 17mmHg OS • External Exam: normal OU • Pupils: No anisocoria and no relative afferent pupillary defect • Motility: Full OU. • Slit lamp exam: megalocornea (>13 mm in diameter) and with anterior mosaic dystrophy. Iris-sutured posterior chamber IOLs (PCIOLs), stable OD, but pseudophacodonesis OS with loose inferior suture evident. • Dilated funduscopic exam: Normal OU Clinical Course: The patient’s iris-sutured IOL had become loose (tilted and de-centered) in his large anterior chamber, despite several sutures that had been placed in the past, resulting now in visual disturbance with movement. FDA and IRB approval was obtained to place an Artisan iris-clip IOL (Ophtec®). He was taken to the OR where his existing IOL was removed using Duet forceps and scissors. The Artisan IOL was placed using enclavation iris forceps. -

Insertion of Aqueous Shunt in Pedicatric Glaucoma
1/29/2018 Challenges of Insertion of Aqueous shunt in paediatric glaucoma Ahmed Elkarmouty MD, FRCS Moorfields Eye Hospital London, UK Classification • Primary Childhood Glaucoma • A- Primary Congenital Glaucoma (PCG) 1: 10,000–18,000 • B- Juvenile Open Angle Glaucoma (JOAG) (5-35 ys,)1 : 50,000. • Secondary Childhood Glaucoma • A- Glaucoma associated with non-acquired ocular anomalies • B- Glaucoma associated with non- acquired systemic disease or syndrome • C- Glaucoma associated with acquired condition • D- Glaucoma following Cataract surgery 1 1/29/2018 Glaucoma associated with non- acquired ocular anomalies • Conditions with predominantly ocular anomalies present at birth which may or may not be associated with systemic signs • Axenfeld Reiger anomaly • Peters anomaly • Ectropion Uvae • Congenital iris hypolplasia • Aniridia • Oculodermal melanocytosis • Posterior polymorphous dystrophy • Microphthalmos • Microcornea • Ectopia Lentis ( et pupillae) • Persistent foetus vasculopathy Glaucoma associated with non- acquired systemic disease or syndrome predominantly associated with known syndrome, systemic anomalies present at birth which may be associated with ocular signs • Down Syndrome • Connective tissue disorder: Marfan syndrome, Weill- Marchesiani syndrome, Stickler syndrome • Metabolic disorder : Homocystenuria, lowe syndrome, Mucoploysacchroidoses • Phacomatoses: Neurofibromatoses, Sturge Weber, Klipple-Trenaunay- weber syndrome, Rubenstein Taybi • Congenital Rubella 2 1/29/2018 Glaucoma associated with acquired condition Conditions -

Expanding the Phenotypic Spectrum of PAX6 Mutations: from Congenital Cataracts to Nystagmus
G C A T T A C G G C A T genes Article Expanding the Phenotypic Spectrum of PAX6 Mutations: From Congenital Cataracts to Nystagmus Maria Nieves-Moreno 1,* , Susana Noval 1 , Jesus Peralta 1, María Palomares-Bralo 2 , Angela del Pozo 3 , Sixto Garcia-Miñaur 4, Fernando Santos-Simarro 4 and Elena Vallespin 5 1 Department of Ophthalmology, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] (S.N.); [email protected] (J.P.) 2 Department of Molecular Developmental Disorders, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] 3 Department of Bioinformatics, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] 4 Department of Clinical Genetics, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] (S.G.-M.); [email protected] (F.S.-S.) 5 Department of Molecular Ophthalmology, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] * Correspondence: [email protected] Abstract: Background: Congenital aniridia is a complex ocular disorder, usually associated with severe visual impairment, generally caused by mutations on the PAX6 gene. The clinical phenotype of PAX6 mutations is highly variable, making the genotype–phenotype correlations difficult to establish. Methods: we describe the phenotype of eight patients from seven unrelated families Citation: Nieves-Moreno, M.; Noval, with confirmed mutations in PAX6, and very different clinical manifestations. -
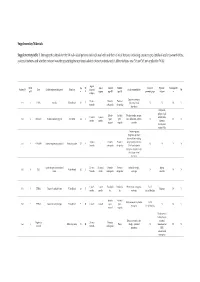
Solved/Unsolved
Supplementary Materials: Supplementary table 1. Demographic details for the 54 individual patients (solved/unsolved) and their clinical features including cataract type, details of ocular co-morbidities, systemic features and whether cataract was the presenting feature (non-isolated cataract patients only). Abbreviations: yes (Y), no (N), not applicable (N/A). Age at Famil Ag M/ Age at Cataract Cataract Cataract Systemic Consanguinit Patient ID Gene Confirmed genetic diagnosis Ethnicity diagnosi Ocular co-morbidities FH y ID e F surgery type RE type LE presenting sign features y s (days) Aniridia, nystagmus, 23 years Posterior Posterior 1-1 1 PAX6 Aniridia White British 25 F - glaucoma, foveal N N N Y 4 months subcapsular subcapsular hypoplasia Cleft palate, epilepsy, high Aphakia Aphakia Macular atrophy, myopia, 7 years 9 7 years 8 arched palate, 2-1 2 COL11A1 Stickler syndrome, type II Not Stated 34 F (post- (post- lens subluxation, vitreous N N N months months flattened surgical) surgical) anomaly maxilla, short stature (5'2ft) Anterior segment dysgenesis, pupillary abnormalities including 12 years Posterior Posterior ectopic pupils, ectropion 3-1 3 CPAMD8 Anterior segment dysgenesis 8 Other, Any other 27 F - N N Y N 5 months subcapsular subcapsular UVAE and irodensis, nystagmus, dysplastic optic discs, large corneal diameters Gyrate atrophy of choroid and 23 years 29 years 1 Posterior Posterior Retinal dystrophy, Bipolar 4-1 4 OAT White British 42 F N N N retina 7 months month subcapsular subcapsular exotropia disorder 1 year 6 1 year -

Journal of Ophthalmology & Clinical Research
ISSN: 2573-9573 Case Report Journal of Ophthalmology & Clinical Research Bilateral Congenital Ectropion Uveae, Anterior Segment Dysgenesis and Aniridia with Microspherophakic Congenital Cataracts and RubeosisIridis Rao Muhammad Arif Khan* and Ashal Kaiser Pal *Corresponding author Rao Muhammad Arif Khan, MCPS, FCPS, FPO, FACS, Pediatric Ophthalmologist, King Edward Medical University, Al-Awali Street, Taif Road, Makkah, Saudi Arabia, Pediatric Ophthalmologist, King Edward Medical University, Tel: 00966560479694; E-mail: [email protected] Makkah, Saudi Arabia Submitted: 02 Apr 2018; Accepted: 12 Apr 2018; Published: 19 Apr 2018 Abstract In recent times, multiple eye diseases have been seen associated with an increase in the rate of Demodex infestation as a possible cause, but in the particular case of dry eye syndrome in patients treated with platelet-rich plasma, this increase in mite may be relevant to guide a more adequate treatment focusing on the elimination of the mite in conjunction with the recovery of the ocular ecology. The demodex mite is a commensal parasite that lives in hair follicles, sebaceous glands and meibomian, which in a high rate of infestation can generate alterations in the ocular area. Performing an adequate diagnosis for the detection of the mite and treatment for its eradication can be effective for the recovery of the normal physiology of the tear film that constitutes a cause of dry eye. Introduction Congenital ectropion uvea is a rare ocular manifestation of neural crest syndrome [1]. It is a non-progressive anomaly characterized by presence of iris pigment epithelium on anterior surface of iris from the pigment ruff [2]. Congenital glaucoma is its common association [3-8]. -

Lid and Lash Conditions
Perth Veterinary Ophthalmology Lid and Lash Conditions Eyelid Diseases The most common eyelid diseases are entropion, ectropion and facial droop. Entropion Entropion means a turning in of the lids. This is a common complaint in young dogs but can sometimes affect older dogs and cats as well. Most cases in young dogs affect the lower lids, but the upper lid can become affected in later life in some breeds such as Cocker Spaniels and Bloodhounds. Entropion Some breeds such as Shar Peis, Chows, Rottweillers and Mastiffs can have very complex entropion leading to defects in both upper and lower lids. A Shar Pei with severe upper and lower lid entropion Entropion is painful and can be potentially blinding. The rolling in of the lid leads to hair coming into contact with the cornea, leading to pain, ulceration and scarring (which can affect vision). In severe cases this can even lead to perforation of the eye. There are many causes of entropion. It can be primary or secondary to other problems affecting the lids (such as ectopic cilia, distichiasis etc. - see below). Some possible causes include the lid being too long, the lid being too tight, instability of the lateral canthus (outer cornea of the eyelids), misdirection of the lateral canthal tendon, brachycephalic anatomy (big eyes and short nose - e.g. Pekingese, Pugs, Shih Tsus, Persian cats etc.), diamond eye defects, loose or too much skin, facial droop etc. Often these cases are referred to a veterinary ophthalmologist for proper assessment and treatment to provide the best outcome. Entropion requires surgical correction. -

Eleventh Edition
SUPPLEMENT TO April 15, 2009 A JOBSON PUBLICATION www.revoptom.com Eleventh Edition Joseph W. Sowka, O.D., FAAO, Dipl. Andrew S. Gurwood, O.D., FAAO, Dipl. Alan G. Kabat, O.D., FAAO Supported by an unrestricted grant from Alcon, Inc. 001_ro0409_handbook 4/2/09 9:42 AM Page 4 TABLE OF CONTENTS Eyelids & Adnexa Conjunctiva & Sclera Cornea Uvea & Glaucoma Viitreous & Retiina Neuro-Ophthalmic Disease Oculosystemic Disease EYELIDS & ADNEXA VITREOUS & RETINA Blow-Out Fracture................................................ 6 Asteroid Hyalosis ................................................33 Acquired Ptosis ................................................... 7 Retinal Arterial Macroaneurysm............................34 Acquired Entropion ............................................. 9 Retinal Emboli.....................................................36 Verruca & Papilloma............................................11 Hypertensive Retinopathy.....................................37 Idiopathic Juxtafoveal Retinal Telangiectasia...........39 CONJUNCTIVA & SCLERA Ocular Ischemic Syndrome...................................40 Scleral Melt ........................................................13 Retinal Artery Occlusion ......................................42 Giant Papillary Conjunctivitis................................14 Conjunctival Lymphoma .......................................15 NEURO-OPHTHALMIC DISEASE Blue Sclera .........................................................17 Dorsal Midbrain Syndrome ..................................45 -

Abnormalities Affecting the Eye As a Whole 2 8 Congenital Corneal
I Editors vi Contributors vii , About the Series viii Preface ix ) Acknowledgments x -t -t Abnormalities Affecting the Eye as a Whole 2 Judith B. Lavrich Anophthalmia 2 Microphthalmia 8 Nanophthaha 12 Typical Coloboma 14 8 Congenital Corneal Opacity 18 Bruce SchnalI and Michael J. Bartiss Sderocornea 18 Birth Trauma: Tears in Descemet's Membrane 20 Ulcer or Lnfection 22 Mucopolysaccharidosis 24 Peters' Anomaly 26 Congenital Hereditary Endothelial Dystrophy 28 Corneal Dermoid 30 Anterior Staphyloma 32 Wilson's Disease (HepatolenticularDegeneration) 34 Herpes Simplex Infection 36 Herpes Simplex Virus Epithelial Dendrite or Ulceration 38 Herpes SimplexVirus Corneal Stromal Disease 40 Herpes Zoster Ophthalmicus 42 Chickenpox 44 Limbal Vernal Keratoconjunctivitis 46 C-3 Glaucoma 48 A& Levin and Anya A. Trumler Primary Congenital or Infantile Glaucoma 48 Juvenile Open-Angle Glaucoma 52 Aphakic Glaucoma 55 Uveitic Glaucoma 58 Sturge-WeberSyndrome 62 m xii CONTENTS Congenital Ectropion Uveae 65 Aniridia 68 Posterior Embryotoxon 70 C- C- 4 Iris Anomalies 72 Michael J.Bartiss and BruceM. Schall Central Pupillary Cysts (Pupillary Margin Epithelial Cysts) 72 Aniridia 74 BrusbJield Spots 76 Ectopia Lentis et Pupillae 78 Heterochromia Iridis 80 Iris Coloboma 82 Iris Stromal Cysts 84 Juvenile Xanthogranuloma 86 Lisch Nodules 88 Melanosis Oculi (Ocular Melanocytosis) 90 Persistent Pupillary Membrane 92 Posterior Synechiae 94 Axenfeld-Rieger Anomaly 96 -5 Lens Anomalies 98 Jonathan H. Salvin and Hillary Gordon Congenital and Developmental Cataracts 98 Ectopia Lentis 102 Anterior Lenticonus 104 Posterior Lenticonus 106 Spherophakia 108 C- 6 Retinal Anomalies 110 Barry N. Wasserman,Anuradha Ganesh, Alex V Levin, Carol L. Shields, Jerry A. Shields, and Alok S. -

An Operation for Congenital Ptosis by George Young
Br J Ophthalmol: first published as 10.1136/bjo.8.6.272 on 1 June 1924. Downloaded from 272 THE BRITISH JOURNAL OF OPHTHALMIOI,OGY added plus lenses (eye being under atropin), J.2, fluently. This made her left eye equal, for distance, to her better eye, which is now getting worse owing to increased bulging, and will probably follow the course of the other one soon. R.V.: 6/36, c. -1.OD sph. + 3.50D cyl. 1550: 6/24 and J.5. Furthermore, it may be noted that the intraocular tension was now normal on the side of the iridectomy, while the right eye was hard, and I submitted it again to pilocarpin and bandage at night. I sent her home for a fortnight to feed up, take malt and cod liver oil and fats, and to take plenty of rest and recuperate. On July 8 the final result was: L.V.: 6/36, c.-5.OD sph. +3.50D cyl. 1600: 6/12 full, and -3.OD sph. and +3.50D cyl. J.2. Soon I shall tattoo the stellate leucoma with an artificial pupil. May 10, 1924. Since writing the above, some ten months ago, affairs have kept steady. There is no bulging of the left cornea or scar. I attempted tattooing at two sittings, and have considerably diminished the glare of the scar, but I refrain from risking the deep tattooing necessary for securing an imitation round black pupil, fearing to do harm. Glasses were prescribed and worn with comfort and great help, the vision being maintained as above. -

Retinitis Pigmentosa Associated with Ectopia Lentis
CLINICOPATHOLOGIC REPORTS, CASE REPORTS, AND SMALL CASE SERIES SECTION EDITOR: W. RICHARD GREEN, MD tion of the oil progressing through the In January 2001, glaucoma sur- Silicone Oil Egressing tube and histopathologic analysis of gery was needed to control elevated Through an Inferiorly the orbital tissue surrounding the ex- intraocular pressure (IOP). The eye Implanted Ahmed Valve truded silicone oil. was aphakic and had total traumatic aniridia. An Ahmed valve was im- Silicone oil use as an adjunct to com- Report of a Case. A 69-year-old white planted inferonasally in an attempt plicated vitreoretinal surgery is be- man lost his left eye to trauma at age to avoid the silicone oil bubble coming more frequent. Refractory 12 years. In September 2000, blunt (Figure 1 and Figure 2). The pa- glaucoma in these patients is com- trauma resulted in a lacerated eye- tient’s IOP responded well initially mon. Isolated reports have men- brow, scleral rupture, uveal prolapse, but rose subsequently to 30 mm Hg. tioned the possibility of silicone oil extrusion of his crystalline lens, reti- A bubble of silicone oil was wrap- migrating and/or obstructing the nal detachment, and suprachoroidal ping the tip of the tube (Figure 3). tube in the anterior chamber of Mol- hemorrhage in his right eye. A limited Silicone oil could be seen migrating teno implants (IOP, Costa Mesa, anterior chamber washout was per- through the Ahmed tube (Figure 4 Calif).1,2 This report describes a case formed at the time of the primary re- and Figure 5) and the bleb over the of intraocular silicone oil egressing pair. -

Congenital Ectopia Lentis - Diagnosis and Treatment
From THE DEPARTMENT OF CLINICAL NEUROSCIENCE, SECTION OF OPHTHALMOLOGY AND VISION, ST. ERIK EYE HOSPITAL Karolinska Institutet, Stockholm, Sweden CONGENITAL ECTOPIA LENTIS - DIAGNOSIS AND TREATMENT Tiina Rysä Konradsen Stockholm 2012 All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by Larserics Digital Print AB. © Tiina Rysä Konradsen, 2012 ISBN 978-91-7457-883-6 ABSTRACT Congenital ectopia lentis (EL) is an ocular condition, which typically causes a high grade of refractive errors, mainly myopia and astigmatism. These might be difficult to compensate for, especially in children, who might develop ametropic amblyopia. Surgery on ectopic lenses has previously been controversial, due to the risk of sight- threatening complications. In paper I we studied retrospectively visual outcomes and complications in children, who were operated for congenital EL, and who had en scleral-fixated capsular tension ring (CTR) and an intra-ocular lens (IOL) implanted at the primary surgery. Thirty-seven eyes of 22 children were included. Visual acuity (VA) improved in all eyes, and only few had persistent amblyopia at the end of the follow-up. A great majority of the eyes had postoperative visual axis opacification (VAO), which was expected, since the posterior capsule was left intact at the primary surgery. Two eyes required secondary suturing for IOL decentration. No eye had any serious complications such as retinal detachment, glaucoma or endophthalmitis. Congenital ectopia lentis is often an indicator of a systemic connective tissue disorder, and Marfan syndrome (MFS) is diagnosed in 70% of the cases. This genetic disorder affects basically all organ systems in the body, EL and dilatation of the ascending aorta being the cardinal signs. -

One-Stage Correction for Blepharophimosis Syndrome
Eye (2008) 22, 380–388 & 2008 Nature Publishing Group All rights reserved 0950-222X/08 $30.00 www.nature.com/eye CLINICAL STUDY One-stage S-Y Wu, L Ma, Y-J Tsai and JZ-C Kuo correction for blepharophimosis syndrome Abstract Introduction Purpose To classify the severity of Blepharophimosis-ptosis-epicanthus inversus blepharophimosis, describe associated (BPES) syndrome is a rare congenital disorder features and their effects on the incidence of and occurs either as an autosomal dominant amblyopia and to recommend guidelines for trait or sporadically.1–6 In 1971, Kohn and surgical treatment and management of surgical Romano2 described the tetrad of this complications. syndrome, which includes blepharophimosis, Methods The case records of 23 patients with blepharoptosis, epicanthus inversus, and blepharophimosis syndrome were examined telecanthus. It is also associated with ocular, retrospectively. Patients’ photographs and lacrimal, nasal, and auricular anomalies.4–6 measurements were reviewed to analyse the There are two subtypes of this syndrome severity of blepharophimosis, surgical proposed by Zlotogora et al:3 type I is more techniques undertaken, surgical outcomes, prevalent and affected female subjects are and complications. Statistical analyses were infertile and type II is transmitted both in performed using paired-sample t-tests to affected male and female subjects.3 evaluate the surgical outcome and Spearman Blepharophimosis syndrome (BPES) has a correlation to examine the influence of great impact on a patient’s functional status and blepharophimosis on the interpalpebral may cause poor visual development.7 Although fissure height (PFH). previous studies describe the demography and Results Eighteen out of 23 (78%) patients aetiology of children with blepharophimosis underwent one-stage surgery before the age syndrome,1–6,8–11 there is a little information in of 5 years.