One-Stage Correction for Blepharophimosis Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Treatment of Congenital Ptosis
13 Review Article Page 1 of 13 Treatment of congenital ptosis Vladimir Kratky1,2^ 1Department of Ophthalmology, Queen’s University, Kingston, Canada; 21st Medical Faculty, Charles University, Prague, Czech Republic Correspondence to: Vladimir Kratky, BSc, MD, FRCSC, DABO. Associate Professor of Ophthalmology, Director of Ophthalmic Plastic and Orbital Surgery, Oculoplastics Fellowship Director, Queen’s University, Kingston, Canada; 1st Medical Faculty, Charles University, Prague, Czech Republic. Email: [email protected]. Abstract: Congenital ptosis is an abnormally low position of the upper eyelid, with respect to the visual axis in the primary gaze. It can be present at birth or manifest itself during the first year of life and can be bilateral or unilateral. Additionally, it may be an isolated finding or part of a constellation of signs of a specific syndrome or systemic associations. Depending on how much it interferes with the visual axis, it may be considered as a functional or a cosmetic condition. In childhood, functional ptosis can lead to deprivation amblyopia and astigmatism and needs to be treated. However, even mild ptosis with normal vision can lead to psychosocial problems and correction is also advised, albeit on a less urgent basis. Although, patching and glasses can be prescribed to treat the amblyopia, the mainstay of management is surgical. There are several types of surgical procedure available depending on the severity and etiology of the droopy eyelid. The first part of this paper will review the different categories of congenital ptosis, including more common associated syndromes. The latter part will briefly cover the different surgical approaches, with emphasis on how to choose the correct condition. -

Novel Anterior Segment Phenotypes Resulting from Forkhead Gene Alterations: Evidence for Cross-Species Conservation of Function
Novel Anterior Segment Phenotypes Resulting from Forkhead Gene Alterations: Evidence for Cross-Species Conservation of Function Ordan J. Lehmann,1 Stephen Tuft,2 Glen Brice,3 Richard Smith,4 Åsa Blixt,5 Rachel Bell,3 Bengt Johansson,6 Tim Jordan,1 Roger A. Hitchings,2 Peng T. Khaw,2 Simon W. M. John,4 Peter Carlsson,5 and Shomi S. Bhattacharya1 PURPOSE. Mutations in murine and human versions of an ances- cause it may affect the clinical management of certain trally related gene usually result in similar phenotypes. How- glaucoma subtypes and lead to excessive treatment. The ever, interspecies differences exist, and in the case of two FOXC1 and Foxe3 data, taken together with the novel ocular forkhead transcription factor genes (FOXC1 and FOXC2), phenotypes of FOXC2 mutations, highlight the remarkable these differences include corneal or anterior segment pheno- cross-species conservation of function among forkhead genes. types, respectively. This study was undertaken to determine (Invest Ophthalmol Vis Sci. 2003;44:2627–2633) DOI:10.1167/ whether such discrepancies provide an opportunity for iden- iovs.02-0609 tifying novel human–murine ocular phenotypes. METHODS. Four pedigrees with early-onset glaucoma pheno- types secondary to segmental chromosomal duplications or ecognition that mutations in orthologous genes frequently deletions encompassing FOXC1 and 18 individuals from 9 Rcause similar phenotypes has allowed the field of compar- FOXC2 mutation pedigrees underwent detailed ocular pheno- ative genetics to contribute to the understanding of human typing. Subsequently, mice with mutations in Foxc1 or a re- disease. As the human, murine, and Drosophila PAX6 mutants lated forkhead gene, Foxe3, were assessed for features of the (aniridia, Small eye, and eyeless) demonstrate, genotypic con- human phenotypes. -

Blepharophimosis, Ptosis, and Epicanthus Inversus Syndrome
Blepharophimosis, ptosis, and epicanthus inversus syndrome Case Report A rare case of adult-onset blepharophimosis, ptosis, and epicanthus inversus syndrome: Case report Mahesha S1, Shruthi Bhimalli2, Manoj Y Bhat2 From 1Chief Medical Officer, 2Fellow in IOL, Department of Cataract and Trauma, Sankara Eye Hospital, Harakere, Shimoga, Karnataka, India Correspondence to: Dr. Shruthi Bhimalli, Department of Cataract and Trauma, Sankara Eye Hospital, Harakere, Shimoga - 577202, Karnataka, India. E-mail: [email protected] Received - 02 June 2019 Initial Review - 24 June 2019 Accepted - 25 July 2019 ABSTRACT Blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES) is a rare genetic condition caused by a mutation in the FOXL2 gene and it is inherited in an autosomal dominant pattern. Identification and diagnosis of BPES syndrome by an ophthalmologist are relatively easy, based on the characteristic ocular manifestations. The most common age group at the time of diagnosis is 4 to 8 years. Here, we present an unusual case of BPES in a patient who presented with the syndrome at the age of 52 years. There is a need for increased awareness about this condition among ophthalmologists as early diagnosis is the key factor in preventing long term complications. Keywords: Blepharophimosis, Epicanthus inversus syndrome, Ptosis. lepharophimosis, ptosis, and epicanthus inversus action, epicanthus inversus and telecanthus (Fig. 1). On acuity syndrome (BPES) is a genetic condition associated testing, his best vision was ‘finger counting close to face’ in his Bwith mutations in the Fork head Box L2 (FOXL2) gene. right eye and ‘finger counting at one meter’ in his left eye. He had The syndrome is inherited in an autosomal dominant pattern, with corneal ectasia with scarring and vascularization (Fig. -

P> Resident’S Day Submission Kelsie Morrison, OD Pediatric Optometry Resident University of Houston College of Optometry
Resident’s Day Submission Kelsie Morrison, OD Pediatric Optometry Resident University of Houston College of Optometry Title: Presumed Blepharophimosis Ptosis Epicanthus Inversus Syndrome (BPES) with Bilateral Type I Duane’s Retraction Syndrome Abstract: Blepharophimosis, ptosis, epicanthus inversus syndrome (BPES) is a rare congenital autosomal dominant condition characterized by bilateral ptosis, telecanthus, epicanthus inversus, and blepharphimosis. The following case highlights the management of ocular manifestations associated with BPES. I. Case History Patient demographics: 7 year old half Caucasian, half Asian male Chief complaint: Annual strabismus evaluation o The patient’s father reported good vision without glasses and non-compliance with spectacle wear. Ocular history: o (+) Bilateral Duane’s retraction syndrome type I o (+) Constant left esotropia with possible strabmismic amblyopia o (+) Telecanthus o (+) Epicanthal folds Medical history: o (+) Unbalanced translocation genetic mutation involving chromosomes 3 and 7 o (+) Developmental delays Systemic surgical history: o (+) Open heart surgery at 1 month old o (+) G-tube at 2 months old o (+) Hand surgery at 1 year old and 1.5 years old o (+) Current G-tube fed secondary to previous ‘failure to thrive’ diagnosis Medications o (+) Hydrocortisone cream o (+) Murolax o No known systemic or ocular medication allergies Family medical and ocular history: o No known family members with chromosomal anomalies Social history: o Attends a special needs school where he is in the -

Insertion of Aqueous Shunt in Pedicatric Glaucoma
1/29/2018 Challenges of Insertion of Aqueous shunt in paediatric glaucoma Ahmed Elkarmouty MD, FRCS Moorfields Eye Hospital London, UK Classification • Primary Childhood Glaucoma • A- Primary Congenital Glaucoma (PCG) 1: 10,000–18,000 • B- Juvenile Open Angle Glaucoma (JOAG) (5-35 ys,)1 : 50,000. • Secondary Childhood Glaucoma • A- Glaucoma associated with non-acquired ocular anomalies • B- Glaucoma associated with non- acquired systemic disease or syndrome • C- Glaucoma associated with acquired condition • D- Glaucoma following Cataract surgery 1 1/29/2018 Glaucoma associated with non- acquired ocular anomalies • Conditions with predominantly ocular anomalies present at birth which may or may not be associated with systemic signs • Axenfeld Reiger anomaly • Peters anomaly • Ectropion Uvae • Congenital iris hypolplasia • Aniridia • Oculodermal melanocytosis • Posterior polymorphous dystrophy • Microphthalmos • Microcornea • Ectopia Lentis ( et pupillae) • Persistent foetus vasculopathy Glaucoma associated with non- acquired systemic disease or syndrome predominantly associated with known syndrome, systemic anomalies present at birth which may be associated with ocular signs • Down Syndrome • Connective tissue disorder: Marfan syndrome, Weill- Marchesiani syndrome, Stickler syndrome • Metabolic disorder : Homocystenuria, lowe syndrome, Mucoploysacchroidoses • Phacomatoses: Neurofibromatoses, Sturge Weber, Klipple-Trenaunay- weber syndrome, Rubenstein Taybi • Congenital Rubella 2 1/29/2018 Glaucoma associated with acquired condition Conditions -

Expanding the Phenotypic Spectrum of PAX6 Mutations: from Congenital Cataracts to Nystagmus
G C A T T A C G G C A T genes Article Expanding the Phenotypic Spectrum of PAX6 Mutations: From Congenital Cataracts to Nystagmus Maria Nieves-Moreno 1,* , Susana Noval 1 , Jesus Peralta 1, María Palomares-Bralo 2 , Angela del Pozo 3 , Sixto Garcia-Miñaur 4, Fernando Santos-Simarro 4 and Elena Vallespin 5 1 Department of Ophthalmology, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] (S.N.); [email protected] (J.P.) 2 Department of Molecular Developmental Disorders, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] 3 Department of Bioinformatics, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] 4 Department of Clinical Genetics, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] (S.G.-M.); [email protected] (F.S.-S.) 5 Department of Molecular Ophthalmology, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] * Correspondence: [email protected] Abstract: Background: Congenital aniridia is a complex ocular disorder, usually associated with severe visual impairment, generally caused by mutations on the PAX6 gene. The clinical phenotype of PAX6 mutations is highly variable, making the genotype–phenotype correlations difficult to establish. Methods: we describe the phenotype of eight patients from seven unrelated families Citation: Nieves-Moreno, M.; Noval, with confirmed mutations in PAX6, and very different clinical manifestations. -
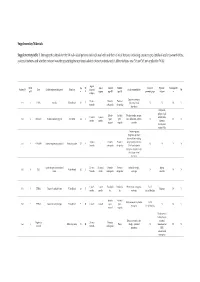
Solved/Unsolved
Supplementary Materials: Supplementary table 1. Demographic details for the 54 individual patients (solved/unsolved) and their clinical features including cataract type, details of ocular co-morbidities, systemic features and whether cataract was the presenting feature (non-isolated cataract patients only). Abbreviations: yes (Y), no (N), not applicable (N/A). Age at Famil Ag M/ Age at Cataract Cataract Cataract Systemic Consanguinit Patient ID Gene Confirmed genetic diagnosis Ethnicity diagnosi Ocular co-morbidities FH y ID e F surgery type RE type LE presenting sign features y s (days) Aniridia, nystagmus, 23 years Posterior Posterior 1-1 1 PAX6 Aniridia White British 25 F - glaucoma, foveal N N N Y 4 months subcapsular subcapsular hypoplasia Cleft palate, epilepsy, high Aphakia Aphakia Macular atrophy, myopia, 7 years 9 7 years 8 arched palate, 2-1 2 COL11A1 Stickler syndrome, type II Not Stated 34 F (post- (post- lens subluxation, vitreous N N N months months flattened surgical) surgical) anomaly maxilla, short stature (5'2ft) Anterior segment dysgenesis, pupillary abnormalities including 12 years Posterior Posterior ectopic pupils, ectropion 3-1 3 CPAMD8 Anterior segment dysgenesis 8 Other, Any other 27 F - N N Y N 5 months subcapsular subcapsular UVAE and irodensis, nystagmus, dysplastic optic discs, large corneal diameters Gyrate atrophy of choroid and 23 years 29 years 1 Posterior Posterior Retinal dystrophy, Bipolar 4-1 4 OAT White British 42 F N N N retina 7 months month subcapsular subcapsular exotropia disorder 1 year 6 1 year -

Journal of Ophthalmology & Clinical Research
ISSN: 2573-9573 Case Report Journal of Ophthalmology & Clinical Research Bilateral Congenital Ectropion Uveae, Anterior Segment Dysgenesis and Aniridia with Microspherophakic Congenital Cataracts and RubeosisIridis Rao Muhammad Arif Khan* and Ashal Kaiser Pal *Corresponding author Rao Muhammad Arif Khan, MCPS, FCPS, FPO, FACS, Pediatric Ophthalmologist, King Edward Medical University, Al-Awali Street, Taif Road, Makkah, Saudi Arabia, Pediatric Ophthalmologist, King Edward Medical University, Tel: 00966560479694; E-mail: [email protected] Makkah, Saudi Arabia Submitted: 02 Apr 2018; Accepted: 12 Apr 2018; Published: 19 Apr 2018 Abstract In recent times, multiple eye diseases have been seen associated with an increase in the rate of Demodex infestation as a possible cause, but in the particular case of dry eye syndrome in patients treated with platelet-rich plasma, this increase in mite may be relevant to guide a more adequate treatment focusing on the elimination of the mite in conjunction with the recovery of the ocular ecology. The demodex mite is a commensal parasite that lives in hair follicles, sebaceous glands and meibomian, which in a high rate of infestation can generate alterations in the ocular area. Performing an adequate diagnosis for the detection of the mite and treatment for its eradication can be effective for the recovery of the normal physiology of the tear film that constitutes a cause of dry eye. Introduction Congenital ectropion uvea is a rare ocular manifestation of neural crest syndrome [1]. It is a non-progressive anomaly characterized by presence of iris pigment epithelium on anterior surface of iris from the pigment ruff [2]. Congenital glaucoma is its common association [3-8]. -

Lid and Lash Conditions
Perth Veterinary Ophthalmology Lid and Lash Conditions Eyelid Diseases The most common eyelid diseases are entropion, ectropion and facial droop. Entropion Entropion means a turning in of the lids. This is a common complaint in young dogs but can sometimes affect older dogs and cats as well. Most cases in young dogs affect the lower lids, but the upper lid can become affected in later life in some breeds such as Cocker Spaniels and Bloodhounds. Entropion Some breeds such as Shar Peis, Chows, Rottweillers and Mastiffs can have very complex entropion leading to defects in both upper and lower lids. A Shar Pei with severe upper and lower lid entropion Entropion is painful and can be potentially blinding. The rolling in of the lid leads to hair coming into contact with the cornea, leading to pain, ulceration and scarring (which can affect vision). In severe cases this can even lead to perforation of the eye. There are many causes of entropion. It can be primary or secondary to other problems affecting the lids (such as ectopic cilia, distichiasis etc. - see below). Some possible causes include the lid being too long, the lid being too tight, instability of the lateral canthus (outer cornea of the eyelids), misdirection of the lateral canthal tendon, brachycephalic anatomy (big eyes and short nose - e.g. Pekingese, Pugs, Shih Tsus, Persian cats etc.), diamond eye defects, loose or too much skin, facial droop etc. Often these cases are referred to a veterinary ophthalmologist for proper assessment and treatment to provide the best outcome. Entropion requires surgical correction. -

Congenital Ocular Anomalies in Newborns: a Practical Atlas
www.jpnim.com Open Access eISSN: 2281-0692 Journal of Pediatric and Neonatal Individualized Medicine 2020;9(2):e090207 doi: 10.7363/090207 Received: 2019 Jul 19; revised: 2019 Jul 23; accepted: 2019 Jul 24; published online: 2020 Sept 04 Mini Atlas Congenital ocular anomalies in newborns: a practical atlas Federico Mecarini1, Vassilios Fanos1,2, Giangiorgio Crisponi1 1Neonatal Intensive Care Unit, Azienda Ospedaliero-Universitaria Cagliari, University of Cagliari, Cagliari, Italy 2Department of Surgery, University of Cagliari, Cagliari, Italy Abstract All newborns should be examined for ocular structural abnormalities, an essential part of the newborn assessment. Early detection of congenital ocular disorders is important to begin appropriate medical or surgical therapy and to prevent visual problems and blindness, which could deeply affect a child’s life. The present review aims to describe the main congenital ocular anomalies in newborns and provide images in order to help the physician in current clinical practice. Keywords Congenital ocular anomalies, newborn, anophthalmia, microphthalmia, aniridia, iris coloboma, glaucoma, blepharoptosis, epibulbar dermoids, eyelid haemangioma, hypertelorism, hypotelorism, ankyloblepharon filiforme adnatum, dacryocystitis, dacryostenosis, blepharophimosis, chemosis, blue sclera, corneal opacity. Corresponding author Federico Mecarini, MD, Neonatal Intensive Care Unit, Azienda Ospedaliero-Universitaria Cagliari, University of Cagliari, Cagliari, Italy; tel.: (+39) 3298343193; e-mail: [email protected]. -

The Management of Congenital Malpositions of Eyelids, Eyes and Orbits
Eye (\988) 2, 207-219 The Management of Congenital Malpositions of Eyelids, Eyes and Orbits S. MORAX AND T. HURBLl Paris Summary Congenital malformations of the eye and its adnexa which are multiple and varied can affect the whole eyeball or any part of it, as well as the orbit, eyelids, lacrimal ducts, extra-ocular muscles and conjunctiva. A classification of these malformations is presented together with the general principles of treatment, age of operating and surgical tactics. The authors give some examples of the anatomo-clinical forms, eyelid malpositions such as entropion, ectropion, ptosis, levator eyelid retraction, medial canthus malposition, congenital eyelid colobomas, and congenital orbital abnormalities (Craniofacial stenosis, orbi tal plagiocephalies, hypertelorism, anophthalmos, microphthalmos and cryptophthalmos) . Congenital malformations of the eye and its as echography, CT-scan and NMR, enzymatic adnexa are multiple and varied. They can work-up or genetic studies (Table I). affect the whole eyeball or any part of it, as Surgical treatment when feasible will well as the orbit, eyelids, lacrimal ducts extra encounter numerous problems; age will play a ocular muscles and conjunctiva. role, choice of a surgical protocol directly From the anatomical point of view, the fol related to the existing complaints, and coop lowing can be considered. eration between several surgical teams Position abnormalities (malpositions) of (ophthalmologic, plastic, cranio-maxillo-fac one or more elements and formation abnor ial and neurosurgical), the ideal being to treat malities (malformations) of the same organs. Some of these abnormalities are limited to Table I The manag ement of cong enital rna/positions one organ and can be subjected to a relatively of eyelid s, eyes and orbits simple and well recognised surgical treat Ocular Findings: ment. -

Ocular Colobomaâ
Eye (2021) 35:2086–2109 https://doi.org/10.1038/s41433-021-01501-5 REVIEW ARTICLE Ocular coloboma—a comprehensive review for the clinician 1,2,3 4 5 5 6 1,2,3,7 Gopal Lingam ● Alok C. Sen ● Vijaya Lingam ● Muna Bhende ● Tapas Ranjan Padhi ● Su Xinyi Received: 7 November 2020 / Revised: 9 February 2021 / Accepted: 1 March 2021 / Published online: 21 March 2021 © The Author(s) 2021. This article is published with open access Abstract Typical ocular coloboma is caused by defective closure of the embryonal fissure. The occurrence of coloboma can be sporadic, hereditary (known or unknown gene defects) or associated with chromosomal abnormalities. Ocular colobomata are more often associated with systemic abnormalities when caused by chromosomal abnormalities. The ocular manifestations vary widely. At one extreme, the eye is hardly recognisable and non-functional—having been compressed by an orbital cyst, while at the other, one finds minimalistic involvement that hardly affects the structure and function of the eye. In the fundus, the variability involves the size of the coloboma (anteroposterior and transverse extent) and the involvement of the optic disc and fovea. The visual acuity is affected when coloboma involves disc and fovea, or is complicated by occurrence of retinal detachment, choroidal neovascular membrane, cataract, amblyopia due to uncorrected refractive errors, etc. While the basic birth anomaly cannot be corrected, most of the complications listed above are correctable to a great 1234567890();,: 1234567890();,: extent. Current day surgical management of coloboma-related retinal detachments has evolved to yield consistently good results. Cataract surgery in these eyes can pose a challenge due to a combination of microphthalmos and relatively hard lenses, resulting in increased risk of intra-operative complications.