The Effects of Exposure to the Environmental Neurotoxicant Manganese on Α-Synuclein and Its Cell-To-Cell Transmission Via Exoso
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

付表 ⅠA 指定を受けた医薬の有効成分 Annex ⅠA Designated
付表ⅠA 指定を受けた医薬の有効成分 Annex ⅠA Designated Pharmaceutical Active Ingredients 号(Sub-heading) 品名 Description 2818.30 アルゲルドラート algeldrate 2833.22 アルスルフ alusulf 2842.10 アルマシラート almasilate 2842.10 シマルドラート simaldrate 2842.90 硫酸アルマドラ ート almadrate sulfate 2842.90 アルマガート almagate 2842.90 カルバルドラード carbaldrate 2842.90 ヒドロタルシト hydrotalcite 2842.90 マガルドラート magaldrate 2843.30 オーラノフィン auranofin 2843.30 金チオグリカニド aurothioglycanide 2843.30 金チオりんご酸ナトリウム sodium aurothiomalate 2843.30 金チオ硫酸ナトリウム sodium aurotiosulfate 2843.90 カルボプラチン carboplatin 2843.90 シスプラチン cisplatin 2843.90 デキソルマプラチン dexormaplatin 2843.90 エンロプラチン enloplatin 2843.90 イプロプラチン iproplatin 2843.90 ロバプラチン lobaplatin 2843.90 ミボプラチン miboplatin 2843.90 ネダプラチン nedaplatin 2843.90 オルマプラチン ormaplatin 2843.90 オキサリプラチン oxaliplatin 2843.90 セブリプラチン sebriplatin 2843.90 スピロプラチン spiroplatin 2843.90 ゼニプラチン zeniplatin 2844.40 アルツモマブ altumomab 2844.40 塩化セシウム(131Cs) cesium (131 Cs) chloride 2844.40 クロルメロドリン(197Hg) chlormerodrin (197 Hg) 2844.40 シアノコバラミン(57Co) cyanocobalamin (57 Co) 2844.40 シアノコバラミン(58Co) cyanocobalamin (58 Co) 2844.40 シアノコバラミン(60Co) cyanocobalamin (60 Co) 2844.40 エチオダイズド油(131I) ethiodized oil (131 I) 2844.40 くえん酸第二鉄(59Fe)注射液 ferric (59 Fe) citrate in 2844.40 フィブリノゲン(125I) fibrinogen (125 I) 2844.40 フルデオキシグルコー ス(18F) fludeoxyglucose ( 18 F) 2844.40 フルオロドパ(18F) fluorodopa (18 F) 2844.40 くえん酸ガリウム(67Ga) gallium (67 Ga) citrate 2844.40 金コロイド(198Au) gold (198 Au), colloidal 2844.40 イオベングアン(131I) iobenguane (131 I) 2844.40 よう化人血清アルブミン(125I) iodinated (125 I) human serum albumin 2844.40 よう化人血清アルブミン(131I) iodinated -

Role of Mesenchymal Stem Cells in Counteracting Oxidative Stress—Related Neurodegeneration
International Journal of Molecular Sciences Review Role of Mesenchymal Stem Cells in Counteracting Oxidative Stress—Related Neurodegeneration Cristina Angeloni 1 , Martina Gatti 2, Cecilia Prata 3,* , Silvana Hrelia 4 and Tullia Maraldi 2 1 School of Pharmacy, University of Camerino, Via Gentile III da Varano, 62032 Camerino, Italy; [email protected] 2 Department of Surgery, Medicine, Dentistry and Morphological Sciences, University of Modena and Reggio Emilia, Via del Pozzo 71, 41124 Modena, Italy; [email protected] (M.G.); [email protected] (T.M.) 3 Department of Pharmacy and Biotechnology, Alma Mater Studiorum—University of Bologna, Via Irnerio 48, 40126 Bologna, Italy 4 Department for Life Quality Studies, Alma Mater Studiorum—University of Bologna, Corso d’Augusto 237, 47921 Rimini, Italy; [email protected] * Correspondence: [email protected] Received: 7 April 2020; Accepted: 4 May 2020; Published: 7 May 2020 Abstract: Neurodegenerative diseases include a variety of pathologies such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, and so forth, which share many common characteristics such as oxidative stress, glycation, abnormal protein deposition, inflammation, and progressive neuronal loss. The last century has witnessed significant research to identify mechanisms and risk factors contributing to the complex etiopathogenesis of neurodegenerative diseases, such as genetic, vascular/metabolic, and lifestyle-related factors, which often co-occur and interact with each other. Apart from several environmental or genetic factors, in recent years, much evidence hints that impairment in redox homeostasis is a common mechanism in different neurological diseases. However, from a pharmacological perspective, oxidative stress is a difficult target, and antioxidants, the only strategy used so far, have been ineffective or even provoked side effects. -

Pharmaceutical Appendix to the Tariff Schedule 2
Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. ABACAVIR 136470-78-5 ACIDUM LIDADRONICUM 63132-38-7 ABAFUNGIN 129639-79-8 ACIDUM SALCAPROZICUM 183990-46-7 ABAMECTIN 65195-55-3 ACIDUM SALCLOBUZICUM 387825-03-8 ABANOQUIL 90402-40-7 ACIFRAN 72420-38-3 ABAPERIDONUM 183849-43-6 ACIPIMOX 51037-30-0 ABARELIX 183552-38-7 ACITAZANOLAST 114607-46-4 ABATACEPTUM 332348-12-6 ACITEMATE 101197-99-3 ABCIXIMAB 143653-53-6 ACITRETIN 55079-83-9 ABECARNIL 111841-85-1 ACIVICIN 42228-92-2 ABETIMUSUM 167362-48-3 ACLANTATE 39633-62-0 ABIRATERONE 154229-19-3 ACLARUBICIN 57576-44-0 ABITESARTAN 137882-98-5 ACLATONIUM NAPADISILATE 55077-30-0 ABLUKAST 96566-25-5 ACODAZOLE 79152-85-5 ABRINEURINUM 178535-93-8 ACOLBIFENUM 182167-02-8 ABUNIDAZOLE 91017-58-2 ACONIAZIDE 13410-86-1 ACADESINE 2627-69-2 ACOTIAMIDUM 185106-16-5 ACAMPROSATE 77337-76-9 -

Marrakesh Agreement Establishing the World Trade Organization
No. 31874 Multilateral Marrakesh Agreement establishing the World Trade Organ ization (with final act, annexes and protocol). Concluded at Marrakesh on 15 April 1994 Authentic texts: English, French and Spanish. Registered by the Director-General of the World Trade Organization, acting on behalf of the Parties, on 1 June 1995. Multilat ral Accord de Marrakech instituant l©Organisation mondiale du commerce (avec acte final, annexes et protocole). Conclu Marrakech le 15 avril 1994 Textes authentiques : anglais, français et espagnol. Enregistré par le Directeur général de l'Organisation mondiale du com merce, agissant au nom des Parties, le 1er juin 1995. Vol. 1867, 1-31874 4_________United Nations — Treaty Series • Nations Unies — Recueil des Traités 1995 Table of contents Table des matières Indice [Volume 1867] FINAL ACT EMBODYING THE RESULTS OF THE URUGUAY ROUND OF MULTILATERAL TRADE NEGOTIATIONS ACTE FINAL REPRENANT LES RESULTATS DES NEGOCIATIONS COMMERCIALES MULTILATERALES DU CYCLE D©URUGUAY ACTA FINAL EN QUE SE INCORPOR N LOS RESULTADOS DE LA RONDA URUGUAY DE NEGOCIACIONES COMERCIALES MULTILATERALES SIGNATURES - SIGNATURES - FIRMAS MINISTERIAL DECISIONS, DECLARATIONS AND UNDERSTANDING DECISIONS, DECLARATIONS ET MEMORANDUM D©ACCORD MINISTERIELS DECISIONES, DECLARACIONES Y ENTEND MIENTO MINISTERIALES MARRAKESH AGREEMENT ESTABLISHING THE WORLD TRADE ORGANIZATION ACCORD DE MARRAKECH INSTITUANT L©ORGANISATION MONDIALE DU COMMERCE ACUERDO DE MARRAKECH POR EL QUE SE ESTABLECE LA ORGANIZACI N MUND1AL DEL COMERCIO ANNEX 1 ANNEXE 1 ANEXO 1 ANNEX -

JAKOVA-THESIS-2016.Pdf (10.00Mb)
THE EFFECTS OF AN AC FIELD ON THE TRANSPORT OF PEPTIDES AND OTHER MOLECULES THROUGH THE α-HEMOLYSIN PORE A Thesis Submitted to the College of Graduate Studies and Research In Partial Fulfillment of the Requirements for the Degree of Master of Science In the Department of Biochemistry University of Saskatchewan, Saskatoon By Elisabet Jakova © Copyright Elisabet Jakova, June 07, 2016. All Rights Reserved. Permission to Use In presenting this thesis in partial fulfillment of the requirements for a Postgraduate degree from the University of Saskatchewan, I agree that the Libraries of this University may make it freely available for inspection. I further agree that permission for copying of this thesis in any manner, in whole or in part, for scholarly purposes may be granted by the professors who supervised my thesis work, or in their absence, by the Head of the Department or the Dean of the College in which my thesis work was done. It is understood that any copying or publication or use of this thesis or parts thereof for financial gain shall not be allowed without my written permission. It is also understood that due to recognition shall be given to me and to the University of Saskatchewan in any scholarly use which may be made of any material in my thesis. Request for permission to copy or make other uses of material in this thesis in whole or in part should be addressed to: Head of the Department of Biochemistry College of Medicine, University of Saskatchewan Saskatoon, SK Canada S7N 5E5 i ABSTRACT This research is based on superimposing an alternating current (AC) onto a direct current (DC) during the nanopore analysis of various peptides, single-stranded DNA, α-synuclein (AS) protein, and AS-drug complexes. -

Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DIX to the HTSUS—Continued
20558 Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DEPARMENT OF THE TREASURY Services, U.S. Customs Service, 1301 TABLE 1.ÐPHARMACEUTICAL APPEN- Constitution Avenue NW, Washington, DIX TO THE HTSUSÐContinued Customs Service D.C. 20229 at (202) 927±1060. CAS No. Pharmaceutical [T.D. 95±33] Dated: April 14, 1995. 52±78±8 ..................... NORETHANDROLONE. A. W. Tennant, 52±86±8 ..................... HALOPERIDOL. Pharmaceutical Tables 1 and 3 of the Director, Office of Laboratories and Scientific 52±88±0 ..................... ATROPINE METHONITRATE. HTSUS 52±90±4 ..................... CYSTEINE. Services. 53±03±2 ..................... PREDNISONE. 53±06±5 ..................... CORTISONE. AGENCY: Customs Service, Department TABLE 1.ÐPHARMACEUTICAL 53±10±1 ..................... HYDROXYDIONE SODIUM SUCCI- of the Treasury. NATE. APPENDIX TO THE HTSUS 53±16±7 ..................... ESTRONE. ACTION: Listing of the products found in 53±18±9 ..................... BIETASERPINE. Table 1 and Table 3 of the CAS No. Pharmaceutical 53±19±0 ..................... MITOTANE. 53±31±6 ..................... MEDIBAZINE. Pharmaceutical Appendix to the N/A ............................. ACTAGARDIN. 53±33±8 ..................... PARAMETHASONE. Harmonized Tariff Schedule of the N/A ............................. ARDACIN. 53±34±9 ..................... FLUPREDNISOLONE. N/A ............................. BICIROMAB. 53±39±4 ..................... OXANDROLONE. United States of America in Chemical N/A ............................. CELUCLORAL. 53±43±0 -
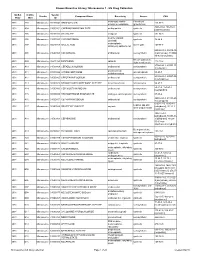
Known Bioactive Library: Microsource 1 - US Drug Collection
Known Bioactive Library: Microsource 1 - US Drug Collection ICCB-L ICCB-L Vendor Vendor Compound Name Bioactivity Source CAS Plate Well ID antifungal, inhibits Penicillium 2091 A03 Microsource 00200046 GRISEOFULVIN 126-07-8 mitosis in metaphase griseofulvum 3505-38-2, 486-16-8 2091 A04 Microsource 01500161 CARBINOXAMINE MALEATE antihistaminic synthetic [carbinoxamine] 2091 A05 Microsource 00200331 SALSALATE analgesic synthetic 552-94-3 muscle relaxant 2091 A06 Microsource 01500162 CARISOPRODOL synthetic 78-44-4 (skeletal) antineoplastic, 2091 A07 Microsource 00210369 GALLIC ACID insect galls 149-91-7 astringent, antibacterial 66592-87-8, 50370-12- 2091 A08 Microsource 01500163 CEFADROXIL antibacterial semisynthetic 2 [anhydrous], 119922- 89-9 [hemihydrate] Rheum palmatum, 2091 A09 Microsource 00211468 DANTHRON cathartic 117-10-2 Xyris semifuscata 27164-46-1, 25953-19- 2091 A10 Microsource 01500164 CEFAZOLIN SODIUM antibacterial semisynthetic 9 [cefazolin] glucocorticoid, 2091 A11 Microsource 00300024 HYDROCORTISONE adrenal glands 50-23-7 antiinflammatory 64485-93-4, 63527-52- 2091 A12 Microsource 01500165 CEFOTAXIME SODIUM antibacterial semisynthetic 6 [cefotaxime] 2091 A13 Microsource 00300029 DESOXYCORTICOSTERONE ACETATE mineralocorticoid adrenocortex 56-47-3 58-71-9, 153-61-7 2091 A14 Microsource 01500166 CEPHALOTHIN SODIUM antibacterial semisynthetic [cephalothin] 2091 A15 Microsource 00300034 TESTOSTERONE PROPIONATE androgen, antineoplastic semisynthetic 57-85-2 24356-60-3, 21593-23- 2091 A16 Microsource 01500167 CEPHAPIRIN SODIUM -

Interaction of the Dopaminergic and Serotonergic Systems in Rat Brain
KUOPION YLIOPISTON JULKAISUJA A. FARMASEUTTISET TIETEET 113 KUOPIO UNIVERSITY PUBLICATIONS A. PHARMACEUTICAL SCIENCES 113 TIINA KÄÄRIÄINEN Interaction of the Dopaminergic and Serotonergic Systems in Rat Brain Studies in Parkinsonian Models and Brain Microdialysis Doctoral dissertation To be presented by permission of the Faculty of Pharmacy of the University of Kuopio for public examination in Mediteknia Auditorium, Mediteknia building, University of Kuopio, on Saturday 13th December 2008, at 1 p.m. Department of Pharmacology and Toxicology Faculty of Pharmacy University of Kuopio JOKA KUOPIO 2008 Distributor: Kuopio University Library P.O. Box 1627 FI-70211 KUOPIO FINLAND Tel. +358 40 355 3430 Fax +358 17 163 410 http://www.uku.fi/kirjasto/julkaisutoiminta/julkmyyn.html Series Editor: Docent Pekka Jarho, Ph.D. Department of Pharmaceutical Chemistry Author’s address: Department of Pharmacology and Toxicology University of Kuopio P.O. Box 1627 FI-70211 KUOPIO Tel. +358 40 355 3776 Fax +358 17 162 424 E-mail: [email protected] Supervisors: Professor Pekka T. Männistö, M.D., Ph.D. Division of Pharmacology and Toxicology Faculty of Pharmacy University of Helsinki Senior assistant Anne Lecklin, Ph.D. Department of Pharmacology and Toxicology University of Kuopio Reviewers: Docent Pekka Rauhala, M.D., Ph.D. Institute of Biomedicine University of Helsinki Docent Seppo Kaakkola, M.D., Ph.D. Department of Neurology Helsinki University Central Hospital Opponent: Professor Raimo K. Tuominen, M.D., Ph.D. Division of Pharmacology and Toxicology Faculty of Pharmacy University of Helsinki ISBN 978-951-27-0851-2 ISBN 978-951-27-1144-4 (PDF) ISSN 1235-0478 Kopijyvä Kuopio 2008 Finland 3 Kääriäinen, Tiina. -

Medicinal Properties of Terpenes Found in Cannabis Sativa and Humulus Lupulus
ACCEPTED MANUSCRIPT Medicinal properties of terpenes found in Cannabis sativa and Humulus lupulus Tarmo Nuutinen 1,2,* 1 Department of Environmental and Biological Sciences, Univerisity of Eastern Finland (UEF) 2 Department of Physics and Mathematics, UEF * Email: [email protected] Abstract Cannabaceae plants Cannabis sativa L. and Humulus lupulus L. are rich in terpenes – both are typically comprised of terpenes as up to 3-5% of the dry-mass of the female inflorescence. Terpenes of cannabis and hops are typically simple mono- and sesquiterpenes derived from two and three isoprene units, respectively. Some terpenes are relatively well known for their potential in biomedicine and have been used in traditional medicine for centuries, while others are yet to be studied in detail. The current, comprehensive review presents terpenes found in cannabis and hops. Terpenes’ medicinal properties are supported by numerous in vitro , animal and clinical trials and show anti-inflammatory, antioxidant, analgesic, anticonvulsive, antidepressant, anxiolytic, anticancer, antitumor, neuroprotective, anti-mutagenic, anti-allergic, antibiotic and anti-diabetic attributes, among others. Because of the very low toxicity, these terpenes are already widely used as food additives and in cosmetic products. Thus, they have been proven safe and well-tolerated. Keywords MANUSCRIPT Monoterpene; sesquiterpene; Cannabis sativa ; Humulus lupulus ; medicine; medicinal property Introduction Plants produce terpenes for interactions with other organisms [1]. Terpenes protect plants against pathogens like mold, fungus and bacteria, and can attract pollinating insects or repel herbivores. Thousands of terpenes have been found across the plantae , but only a small percentage of all terpenes have been identified [2]. Despite the diversity of the known terpenes, some are concentrated in certain phyla or families such as Cannabaceae . -

(12) United States Patent (Lo) Patent No.: �US 8,480,637 B2
111111111111111111111111111111111111111111111111111111111111111111111111 (12) United States Patent (lo) Patent No.: US 8,480,637 B2 Ferrari et al. (45) Date of Patent : Jul. 9, 2013 (54) NANOCHANNELED DEVICE AND RELATED USPC .................. 604/264; 907/700, 902, 904, 906 METHODS See application file for complete search history. (75) Inventors: Mauro Ferrari, Houston, TX (US); (56) References Cited Xuewu Liu, Sugar Land, TX (US); Alessandro Grattoni, Houston, TX U.S. PATENT DOCUMENTS (US); Daniel Fine, Austin, TX (US); 5,651,900 A 7/1997 Keller et al . .................... 216/56 Randy Goodall, Austin, TX (US); 5,728,396 A 3/1998 Peery et al . ................... 424/422 Sharath Hosali, Austin, TX (US); Ryan 5,770,076 A 6/1998 Chu et al ....................... 210/490 5,798,042 A 8/1998 Chu et al ....................... 210/490 Medema, Pflugerville, TX (US); Lee 5,893,974 A 4/1999 Keller et al . .................. 510/483 Hudson, Elgin, TX (US) 5,938,923 A 8/1999 Tu et al . ........................ 210/490 5,948,255 A * 9/1999 Keller et al . ............. 210/321.84 (73) Assignees: The Board of Regents of the University 5,985,164 A 11/1999 Chu et al ......................... 516/41 of Texas System, Austin, TX (US); The 5,985,328 A 11/1999 Chu et al ....................... 424/489 Ohio State University Research (Continued) Foundation, Columbus, OH (US) FOREIGN PATENT DOCUMENTS (*) Notice: Subject to any disclaimer, the term of this WO WO 2004/036623 4/2004 WO WO 2006/113860 10/2006 patent is extended or adjusted under 35 WO WO 2009/149362 12/2009 U.S.C. 154(b) by 612 days. -
Chemical Structure-Related Drug-Like Criteria of Global Approved Drugs
Molecules 2016, 21, 75; doi:10.3390/molecules21010075 S1 of S110 Supplementary Materials: Chemical Structure-Related Drug-Like Criteria of Global Approved Drugs Fei Mao 1, Wei Ni 1, Xiang Xu 1, Hui Wang 1, Jing Wang 1, Min Ji 1 and Jian Li * Table S1. Common names, indications, CAS Registry Numbers and molecular formulas of 6891 approved drugs. Common Name Indication CAS Number Oral Molecular Formula Abacavir Antiviral 136470-78-5 Y C14H18N6O Abafungin Antifungal 129639-79-8 C21H22N4OS Abamectin Component B1a Anthelminithic 65195-55-3 C48H72O14 Abamectin Component B1b Anthelminithic 65195-56-4 C47H70O14 Abanoquil Adrenergic 90402-40-7 C22H25N3O4 Abaperidone Antipsychotic 183849-43-6 C25H25FN2O5 Abecarnil Anxiolytic 111841-85-1 Y C24H24N2O4 Abiraterone Antineoplastic 154229-19-3 Y C24H31NO Abitesartan Antihypertensive 137882-98-5 C26H31N5O3 Ablukast Bronchodilator 96566-25-5 C28H34O8 Abunidazole Antifungal 91017-58-2 C15H19N3O4 Acadesine Cardiotonic 2627-69-2 Y C9H14N4O5 Acamprosate Alcohol Deterrant 77337-76-9 Y C5H11NO4S Acaprazine Nootropic 55485-20-6 Y C15H21Cl2N3O Acarbose Antidiabetic 56180-94-0 Y C25H43NO18 Acebrochol Steroid 514-50-1 C29H48Br2O2 Acebutolol Antihypertensive 37517-30-9 Y C18H28N2O4 Acecainide Antiarrhythmic 32795-44-1 Y C15H23N3O2 Acecarbromal Sedative 77-66-7 Y C9H15BrN2O3 Aceclidine Cholinergic 827-61-2 C9H15NO2 Aceclofenac Antiinflammatory 89796-99-6 Y C16H13Cl2NO4 Acedapsone Antibiotic 77-46-3 C16H16N2O4S Acediasulfone Sodium Antibiotic 80-03-5 C14H14N2O4S Acedoben Nootropic 556-08-1 C9H9NO3 Acefluranol Steroid -

Dopaminergic Neurons Regenerate Following Chemogenetic Ablation in the Olfactory Bulb of Adult Zebrafish (Danio Rerio)
www.nature.com/scientificreports OPEN Dopaminergic neurons regenerate following chemogenetic ablation in the olfactory bulb of adult Zebrafsh (Danio rerio) Rafael Godoy1, Khang Hua1, Michael Kalyn1, Victoria‑Marie Cusson1, Hymie Anisman2 & Marc Ekker1* Adult zebrafsh have the ability to regenerate cells of the central nervous system. However, few neuronal regeneration studies in adult zebrafsh addressed their ability to regenerate specifc types of neurons following cell specifc ablation. We show here that treatment of transgenic Tg(dat:CFP- NTR) adult zebrafsh with the prodrug metronidazole (Mtz) according to our administration regimen predominantly ablates dopamine (DA) neurons within the olfactory bulb (OB) of adult fsh. Loss of DA neurons was accompanied by an impaired olfaction phenotype, as early as 1-week post-treatment, in which fsh were unable to sense the presence of the repulsive stimulus cadaverine. The olfactory impairment was reversed within 45 days and coincided with the recovery of DA neuron counts in the OB. A multi-label pulse-chase analysis with BrdU and EdU over the frst seventeen days-post Mtz exposure showed that newly formed DA neurons were recruited within the frst nine days following exposure and led to functional and morphological recovery of the OB. Over the last decade, the zebrafsh has emerged as a strong model for the study of neurogenesis and brain regen- eration as previously reviewed 1–3. Unlike mammals, which have two main proliferation zones—the subgranular and subventricular zones 4,5, the zebrafsh holds 16 diferent zones of proliferation in the brain 6,7. Tis ability to hold numerous cells in a proliferating stage allows for the recruitment of stem-progenitor cells to multiply and migrate following damage to brain tissue in order to allow cells to then remodel the brain environment in a regenerative fashion8,9.