Rare Coding Variants in Five DNA Damage Repair Genes Associate
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DNA Topoisomerases and Cancer Topoisomerases and TOP Genes in Humans Humans Vs
DNA Topoisomerases Review DNA Topoisomerases And Cancer Topoisomerases and TOP Genes in Humans Humans vs. Escherichia Coli Topoisomerase differences Comparisons Topoisomerase and genomes Top 1 Top1 and Top2 differences Relaxation of DNA Top1 DNA supercoiling DNA supercoiling In the context of chromatin, where the rotation of DNA is constrained, DNA supercoiling (over- and under-twisting and writhe) is readily generated. TOP1 and TOP1mt remove supercoiling by DNA untwisting, acting as “swivelases”, whereas TOP2a and TOP2b remove writhe, acting as “writhases” at DNA crossovers (see TOP2 section). Here are some basic facts concerning DNA supercoiling that are relevant to topoisomerase activity: • Positive supercoiling (Sc+) tightens the DNA helix whereas negative supercoiling (Sc-) facilitates the opening of the duplex and the generation of single-stranded segments. • Nucleosome formation and disassembly absorbs and releases Sc-, respectively. • Polymerases generate Sc+ ahead and Sc- behind their tracks. • Excess of Sc+ arrests DNA tracking enzymes (helicases and polymerases), suppresses transcription elongation and initiation, and destabilizes nucleosomes. • Sc- facilitates DNA melting during the initiation of replication and transcription, D-loop formation and homologous recombination and nucleosome formation. • Excess of Sc- favors the formation of alternative DNA structures (R-loops, guanine quadruplexes, right-handed DNA (Z-DNA), plectonemic structures), which then absorb Sc- upon their formation and attract regulatory proteins. The -

Type IA Topoisomerases As Targets for Infectious Disease Treatments
microorganisms Review Type IA Topoisomerases as Targets for Infectious Disease Treatments Ahmed Seddek 1,2 , Thirunavukkarasu Annamalai 1 and Yuk-Ching Tse-Dinh 1,2,* 1 Department of Chemistry and Biochemistry, Florida International University, Miami, FL 33199, USA; asedd001@fiu.edu (A.S.); athiruna@fiu.edu (T.A.) 2 Biomolecular Sciences Institute, Florida International University, Miami, FL 33199, USA * Correspondence: ytsedinh@fiu.edu; Tel.: +1-305-348-4956 Abstract: Infectious diseases are one of the main causes of death all over the world, with antimi- crobial resistance presenting a great challenge. New antibiotics need to be developed to provide therapeutic treatment options, requiring novel drug targets to be identified and pursued. DNA topoi- somerases control the topology of DNA via DNA cleavage–rejoining coupled to DNA strand passage. The change in DNA topological features must be controlled in vital processes including DNA repli- cation, transcription, and DNA repair. Type IIA topoisomerases are well established targets for antibiotics. In this review, type IA topoisomerases in bacteria are discussed as potential targets for new antibiotics. In certain bacterial pathogens, topoisomerase I is the only type IA topoisomerase present, which makes it a valuable antibiotic target. This review will summarize recent attempts that have been made to identify inhibitors of bacterial topoisomerase I as potential leads for antibiotics and use of these inhibitors as molecular probes in cellular studies. Crystal structures of inhibitor–enzyme complexes and more in-depth knowledge of their mechanisms of actions will help to establish the structure–activity relationship of potential drug leads and develop potent and selective therapeutics that can aid in combating the drug resistant bacterial infections that threaten public health. -
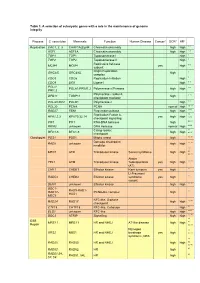
Table 1. a Selection of Eukaryotic Genes with a Role in the Maintenance of Genome Integrity
Table 1. A selection of eukaryotic genes with a role in the maintenance of genome integrity Process S. cerevisiae Mammals Function Human Disease Cancer* GCR* HR* Replication CAC1, 2, 3 CHAF1A,B,p48 Chromatin assembly high high 1, 2 1, 2 ASF1 ASF1A Chromatin assembly high high 3 TOP1 TOP1 Topoisomerase I high 3 TOP2 TOP2 Topoisomerase II high Replicative helicase 4, 5 MCM4 MCM4 yes high subunit Origin Replication 6 ORC3/5 ORC3/6L high complex 7 CDC6 CDC6 Replication Initiation high 7, 8 CDC9 LIG1 Ligase I high POL1/ 7-10 POLA1/PRIM1,2 Polymerase α/Primase high high PRI1,2 Polymerase ε subunit, 6, 11 DPB11 TOBP11 high checkpoint mediator POL3/CDC2 POLD1 Polymerase δ high 7, 8 POL30 PCNA PCNA normal high 12, 13 RAD27 FEN1 Flap endonuclease high high 14-16 Replication Factor A, 14, RFA1,2,3 RPA70,32,14 yes high high 17-19 checkpoint signalling PIF1 PIF1 RNA-DNA helicase high 20, 21 RRM3 unknown DNA Helicase normal high 22-26 Clamp loader, 11, RFC1-5 RFC1-5 high high 20, 27 checkpoint Checkpoint PDS1 PDS1 Mitotic arrest high 11, 28 Damage checkpoint 11, 29 RAD9 unknown high high mediator 11, MEC1 ATR Transducer kinase Seckel syndrome high high 28, 30, 31 Ataxia TEL1 ATM Transducer kinase Telangiectasia yes high high 11, 28 (AT) CHK1 CHEK1 Effector kinase Rare tumours yes high 11 Li-Fraumeni RAD53 CHEK2 Effector kinase syndrome yes high 11 variant DUN1 unknown Effector kinase high high 11, 32 DDC1- RAD9-RAD1- 11 RAD17- PCNA-like complex high HUS1 MEC3 RFC-like, S-phase 11, 33 RAD24 RAD17 high high checkponit 34 CTF18 CHTF18 RFC-like, Cohesion -

Identification of Gastric Cancer–Related Genes Using a Cdna Microarray Containing Novel Expressed Sequence Tags Expressed in Gastric Cancer Cells
Vol. 11, 473–482, January 15, 2005 Clinical Cancer Research 473 Identification of Gastric Cancer–Related Genes Using a cDNA Microarray Containing Novel Expressed Sequence Tags Expressed in Gastric Cancer Cells Jeong-Min Kim,1,5 Ho-Yong Sohn,4 overexpressed in z68% of tissues and the MT2A gene Sun Young Yoon,1 Jung-Hwa Oh,1 Jin Ok Yang,1 was down-expressed in 72% of the tissues. Western blotting and immunohistochemical analyses for CDC20 and SKB1 Joo Heon Kim,2 Kyu Sang Song,3 Seung-Moo Rho,2 1 1 5 showed overexpression and localization changes of the Hyan Sook Yoo, Yong Sung Kim, Jong-Guk Kim, corresponding protein in human gastric cancer tissues. 1 and Nam-Soon Kim Conclusions: Novel genes that are related to human 1Genome Research Center, Korea Research Institute of Bioscience and gastric cancer were identified using cDNA microarray Biotechnology; 2Department of Pathology, Eulji University School of 3 developed in our laboratory. In particular, CDC20 and Medicine; and Department of Pathology, College of Medicine, MT2A represent a potential biomarker of human gastric Chungnam National University, Daejeon, Korea; 4Department of Food and Nutrition, Andong National University, Andong, Korea; and cancer. These newly identified genes should provide a 5Department of Microbiology, College of Natural Sciences, Kyungpook valuable resource for understanding the molecular mecha- National University, Daegu, Korea nism associated with tumorigenesis of gastric carcinogenesis and for the discovery of potential diagnostic markers of gastric cancer. ABSTRACT Purpose: Gastric cancer is one of the most frequently INTRODUCTION diagnosed malignancies in the world, especially in Korea and Japan. -

Two Closely Related Recq Helicases Have Antagonistic Roles in Homologous Recombination and DNA Repair in Arabidopsis Thaliana
Two closely related RecQ helicases have antagonistic roles in homologous recombination and DNA repair in Arabidopsis thaliana Frank Hartung, Stefanie Suer, and Holger Puchta* Botany II, University Karlsruhe, Kaiserstrasse 12, D-76128 Karlsruhe, Germany Edited by Nina Fedoroff, U.S. Department of State, Washington, DC, and approved October 4, 2007 (received for review June 26, 2007) RecQ helicases are involved in the processing of DNA structures RMI1 protein was characterized in mammals and yeast (10, 18, arising during replication, recombination, and repair throughout all 19). Moreover, in the lower eukaryotes S. cerevisiae and Schizo- kingdoms of life. Mutations of different RecQ homologues are re- saccharomyces pombe deletion of their respective RecQ homo- sponsible for severe human diseases, such as Blooms (BLM) or Werner logue leads to partial suppression of the severe phenotypes (WRN) syndrome. The loss of RecQ function is often accompanied by caused by mutation of the TOP3 gene (14, 20–22). hyperrecombination caused by a lack of crossover suppression. In the Several RecQ helicase mutants are synthetically lethal in a Arabidopsis genome seven different RecQ genes are present. Two of combination with mutations in the endonuclease MUS81 (9, 23, them (AtRECQ4A and 4B) arose because of a recent duplication and 24). This inviability of the double mutants is most probably because are still nearly 70% identical on a protein level. Knockout of these both proteins act in parallel pathways resolving aberrant DNA genes leads to antagonistic phenotypes: the RECQ4A mutant shows structures arising during replication. When both genes are mutated, sensitivity to DNA-damaging agents, enhanced homologous recom- these structures accumulate, leading to cell death (9, 23). -

(12) United States Patent (10) Patent No.: US 8,148,129 B2 Frankel Et Al
US008148129B2 (12) United States Patent (10) Patent No.: US 8,148,129 B2 Frankel et al. (45) Date of Patent: Apr. 3, 2012 (54) GENERATION OF POTENT DOMINANT 6,824,978 B1 1 1/2004 Cox, III et al. NEGATIVE TRANSCRIPTIONAL 6,933,113 B2 8, 2005 Case et al. 6,979,539 B2 12/2005 Cox, III et al. INHIBITORS 7,013,219 B2 3/2006 Case et al. 7,070,934 B2 7/2006 Cox, III et al. (75) Inventors: Alan Frankel, Mill Valley, CA (US); 7,163,824 B2 1/2007 Cox, III et al. Robert Nakamura, San Francisco, CA 7,220,719 B2 5/2007 Case et al. (US); Chandreyee Das, Brookline, MA 7,235,354 B2 6/2007 Case et al. 7,262,054 B2 8/2007 Jamieson et al. (US); Ivan D’Orso, San Francisco, CA 7,273,923 B2 9/2007 Jamieson et al. (US); Jocelyn Grunwell, San Mateo, 2003, OO82552 A1* 5, 2003 Wolffe et al. ..................... 435/6 CA (US) (73) Assignee: The Regents of the University of OTHER PUBLICATIONS California, Oakland, CA (US) Cramer et al., Coupling of Transcription with Alternative Splicing: RNA Pol II Promoters Modulate SF2. ASF and 9G8 Effects on an (*) Notice: Subject to any disclaimer, the term of this Exonic Splicing Enhancer, Molecular Cell, 1999, 4:251-258.* patent is extended or adjusted under 35 Cama-Carvalho et al., Nucleocytoplasmic shuttling of heterodimeric U.S.C. 154(b) by 806 days. splicing factor U2AF, JBC. Published on Dec. 15, 2000 as Manu script M008759200.* (21) Appl. No.: 11/765,592 Rosonina et al., Gene Expression: The Close Coupling of Transcrip tion and Splicing, Current Biology, vol. -

SUMO-Targeted Ubiquitin Ligases and Their Functions in Maintaining Genome Stability
International Journal of Molecular Sciences Review SUMO-Targeted Ubiquitin Ligases and Their Functions in Maintaining Genome Stability Ya-Chu Chang † , Marissa K. Oram † and Anja-Katrin Bielinsky * Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minnesota, MN 55455, USA; [email protected] (Y.-C.C.); [email protected] (M.K.O.) * Correspondence: [email protected]; Tel.: +1-612-624-2469 † These authors contributed equally. Abstract: Small ubiquitin-like modifier (SUMO)-targeted E3 ubiquitin ligases (STUbLs) are special- ized enzymes that recognize SUMOylated proteins and attach ubiquitin to them. They therefore connect the cellular SUMOylation and ubiquitination circuits. STUbLs participate in diverse molec- ular processes that span cell cycle regulated events, including DNA repair, replication, mitosis, and transcription. They operate during unperturbed conditions and in response to challenges, such as genotoxic stress. These E3 ubiquitin ligases modify their target substrates by catalyzing ubiquitin chains that form different linkages, resulting in proteolytic or non-proteolytic outcomes. Often, STUbLs function in compartmentalized environments, such as the nuclear envelope or kine- tochore, and actively aid in nuclear relocalization of damaged DNA and stalled replication forks to promote DNA repair or fork restart. Furthermore, STUbLs reside in the same vicinity as SUMO proteases and deubiquitinases (DUBs), providing spatiotemporal control of their targets. In this review, we focus on the molecular mechanisms by which STUbLs help to maintain genome stability Citation: Chang, Y.-C.; Oram, M.K.; across different species. Bielinsky, A.-K. SUMO-Targeted Ubiquitin Ligases and Their Keywords: genome stability; STUbL; SUMO; ubiquitin; Slx5/Slx8; RNF4; RNF111 Functions in Maintaining Genome Stability. -

Characterization of Recombinant Human DNA Topoisomerase Iiiƒї
J Biochem. 127. 1109-1113(2000) Characterization of Recombinant Human DNA Topoisomerase IIIƒ¿ Activity Expressed in Yeast1 Nika Hotoda2 and Ryo Hanai3 Department of Chemistry , College of Science,Rikkyo (St. Paul's) Unioersity,3-34-1 Nishi-Ikebukure, Toshirna-ku, Tokyo171-8501 Received January 28.2000; accepted April 3, 2000 Recombinant human DNA topoisomerase IIIƒ¿ was expressed in mutant yeast cells devoid of both topoisomerases I and III, and the gene product was partially purified. The activity of the protein in supercoil removal was found to be limited and also bipha sic: in the first phase it processively changed the linking-number of hypernegatively supercoiled DNA, but only to the superhelicity of a regular negative supercoil; in the second phase the enzyme relaxed the DNA further, but only slightly and slowly. The optimal solution conditions for the enzyme activity were found to be physiological. The assay results with a truncation mutant showed that the C-terminal 334 amino acids are unnecessary for the activity, suggesting that this region, and perhaps the entire protein, is involved in a function other than supercoil removal. Key words: DNA supereoiling, DNA topoisomerase. Mammalian DNA topoisomerase III (TOP3 gene product), merase I (for a review, 14), but absent in yeast topoiso a type-IA topoisomerase, was first identified in man (I), merase III. Although bacterial topoisomerase I can relax and has been shown to be essential for embryonic develop negatively supercoiled DNA nearly completely, eukaryotic ment in the mouse (2). More recently, a second TOP3 gene type-IA topoisomerases studied so far (1, 4, 12), including was identified (3-5), and the first gene has been renamed the yeast enzyme, appear to remove negative supercoils TOP3ƒ¿ and the second one designated as TOP3ƒÀ (2). -

Topoisomerase 3A and RMI1 Suppress Somatic Crossovers and Are Essential for Resolution of Meiotic Recombination Intermediates in Arabidopsis Thaliana
Topoisomerase 3a and RMI1 Suppress Somatic Crossovers and Are Essential for Resolution of Meiotic Recombination Intermediates in Arabidopsis thaliana Frank Hartung, Stefanie Suer, Alexander Knoll, Rebecca Wurz-Wildersinn, Holger Puchta* Botany II, University of Karlsruhe, Karlsruhe, Germany Abstract Topoisomerases are enzymes with crucial functions in DNA metabolism. They are ubiquitously present in prokaryotes and eukaryotes and modify the steady-state level of DNA supercoiling. Biochemical analyses indicate that Topoisomerase 3a (TOP3a) functions together with a RecQ DNA helicase and a third partner, RMI1/BLAP75, in the resolution step of homologous recombination in a process called Holliday Junction dissolution in eukaryotes. Apart from that, little is known about the role of TOP3a in higher eukaryotes, as knockout mutants show early lethality or strong developmental defects. Using a hypomorphic insertion mutant of Arabidopsis thaliana (top3a-2), which is viable but completely sterile, we were able to define three different functions of the protein in mitosis and meiosis. The top3a-2 line exhibits fragmented chromosomes during mitosis and sensitivity to camptothecin, suggesting an important role in chromosome segregation partly overlapping with that of type IB topoisomerases. Furthermore, AtTOP3a, together with AtRECQ4A and AtRMI1, is involved in the suppression of crossover recombination in somatic cells as well as DNA repair in both mammals and A. thaliana. Surprisingly, AtTOP3a is also essential for meiosis. The phenotype of chromosome fragmentation, bridges, and telophase I arrest can be suppressed by AtSPO11 and AtRAD51 mutations, indicating that the protein is required for the resolution of recombination intermediates. As Atrmi1 mutants have a similar meiotic phenotype to Attop3a mutants, both proteins seem to be involved in a mechanism safeguarding the entangling of homologous chromosomes during meiosis. -

From Microscopy to Nanoscopy: Defining an Arabidopsis Thaliana Meiotic Atlas at the Nanometer Scale
REVIEW published: 18 May 2021 doi: 10.3389/fpls.2021.672914 From Microscopy to Nanoscopy: Defining an Arabidopsis thaliana Meiotic Atlas at the Nanometer Scale Jason Sims* †, Peter Schlögelhofer † and Marie-Therese Kurzbauer * † Department of Chromosome Biology, Max Perutz Labs, University of Vienna, Vienna BioCenter, Vienna, Austria Visualization of meiotic chromosomes and the proteins involved in meiotic recombination have become essential to study meiosis in many systems including the model plant Edited by: Arabidopsis thaliana. Recent advances in super-resolution technologies changed how Christophe Lambing, University of Cambridge, microscopic images are acquired and analyzed. New technologies enable observation of United Kingdom cells and nuclei at a nanometer scale and hold great promise to the field since they allow Reviewed by: observing complex meiotic molecular processes with unprecedented detail. Here, Isabelle Colas, The James Hutton Institute, we provide an overview of classical and advanced sample preparation and microscopy United Kingdom techniques with an updated Arabidopsis meiotic atlas based on super-resolution Alice R. Darbyshire, microscopy. We review different techniques, focusing on stimulated emission depletion University of Birmingham, United Kingdom (STED) nanoscopy, to offer researchers guidance for selecting the optimal protocol and *Correspondence: equipment to address their scientific question. Jason Sims [email protected] Keywords: Arabidopsis, meiosis, cytology, super-resolution microscopy, immunofluorescence Marie-Therese Kurzbauer [email protected] †ORCID: MEIOSIS Jason Sims orcid.org/0000-0003-4235-2488 Meiosis is a specialized cell division and the basis for genetic diversity through sexual reproduction. Peter Schlögelhofer Understanding its molecular mechanisms and involved factors is therefore essential for human orcid.org/0000-0002-0909-3587 health and fertility and, importantly, for plant breeding and food security. -

Maintenance of Genome Integrity and Active Homologous Recombination
Choi et al. Experimental & Molecular Medicine (2020) 52:1220–1229 https://doi.org/10.1038/s12276-020-0481-2 Experimental & Molecular Medicine REVIEW ARTICLE Open Access Maintenance of genome integrity and active homologous recombination in embryonic stem cells Eui-Hwan Choi1, Seobin Yoon1, Young Eun Koh1, Young-Jin Seo1 and Keun Pil Kim1 Abstract Embryonic stem cells (ESCs) possess specific gene expression patterns that confer the ability to proliferate indefinitely and enable pluripotency, which allows ESCs to differentiate into diverse cell types in response to developmental signals. Compared to differentiated cells, ESCs harbor an elevated level of homologous recombination (HR)-related proteins and exhibit exceptional cell cycle control, characterized by a high proliferation rate and a prolonged S phase. HR is involved in several aspects of chromosome maintenance. For instance, HR repairs impaired chromosomes and prevents the collapse of DNA replication forks during cell proliferation. Thus, HR is essential for the maintenance of genomic integrity and prevents cellular dysregulation and lethal events. In addition, abundant HR proteins in the prolonged S phase can efficiently protect ESCs from external damages and protect against genomic instability caused by DNA breaks, facilitating rapid and accurate DNA break repair following chromosome duplication. The maintenance of genome integrity is key to preserving the functions of ESCs and reducing the risks of cancer development, cell cycle arrest, and abnormal replication. Here, we review the fundamental links between the stem cell-specific HR process and DNA damage response as well as the different strategies employed by ESCs to maintain genomic integrity. 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; Introduction lesions is addressed by a tightly controlled DNA damage Embryonic stem cells (ESCs), derived from inner cell response (DDR) system. -
Identification of Human Haploinsufficient Genes and Their Genomic Proximity to Segmental Duplications
European Journal of Human Genetics (2008) 16, 1350–1357 & 2008 Macmillan Publishers Limited All rights reserved 1018-4813/08 $32.00 www.nature.com/ejhg ARTICLE Identification of human haploinsufficient genes and their genomic proximity to segmental duplications Vinh T Dang1,2,3, Karin S Kassahn1,2,3, Andre´s Esteban Marcos1,2 and Mark A Ragan*,1,2 1ARC Centre of Excellence in Bioinformatics, Brisbane, Queensland, Australia; 2Institute for Molecular Bioscience, The University of Queensland, Brisbane, Queensland, Australia Despite the significance of haploinsufficiency in human disease, no systematic study has been reported into the types of genes that are haploinsufficient in human, or into the mechanisms that commonly lead to their deletion and to the expression of the haploinsufficient phenotype. We have applied a rigorous text-searching and database-mining strategy to extract, as comprehensively as possible, from PubMed and OMIM an annotated list of currently known human haploinsufficient genes, including their functions and associated diseases. Gene-set enrichment analysis shows that genes-encoding transcription factors, and genes that function in development, the cell cycle, and nucleic acid metabolism are overrepresented among haploinsufficient genes in human. Many of the phenotypes associated with loss-of-function or deletion of one copy of a haploinsufficient gene describe mental retardation, developmental or metabolic disorders, or tumourigenesis. We also found that haploinsufficient genes are less likely than the complete set of human genes to be situated between pairs of segmental duplications (SDs) that are in close proximity to each other on the same chromosome. Given that SDs can initiate non-allelic homologous recombination (NAHR) and the deletion of adjacent genomic regions, this suggests that the location of haploinsufficient genes between SD pairs, from whence they may suffer intra-genomic rearrangement and loss, is selectively disadvantageous.