Uncorrected Proof
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gasket Chemical Services Guide
Gasket Chemical Services Guide Revision: GSG-100 6490 Rev.(AA) • The information contained herein is general in nature and recommendations are valid only for Victaulic compounds. • Gasket compatibility is dependent upon a number of factors. Suitability for a particular application must be determined by a competent individual familiar with system-specific conditions. • Victaulic offers no warranties, expressed or implied, of a product in any application. Contact your Victaulic sales representative to ensure the best gasket is selected for a particular service. Failure to follow these instructions could cause system failure, resulting in serious personal injury and property damage. Rating Code Key 1 Most Applications 2 Limited Applications 3 Restricted Applications (Nitrile) (EPDM) Grade E (Silicone) GRADE L GRADE T GRADE A GRADE V GRADE O GRADE M (Neoprene) GRADE M2 --- Insufficient Data (White Nitrile) GRADE CHP-2 (Epichlorohydrin) (Fluoroelastomer) (Fluoroelastomer) (Halogenated Butyl) (Hydrogenated Nitrile) Chemical GRADE ST / H Abietic Acid --- --- --- --- --- --- --- --- --- --- Acetaldehyde 2 3 3 3 3 --- --- 2 --- 3 Acetamide 1 1 1 1 2 --- --- 2 --- 3 Acetanilide 1 3 3 3 1 --- --- 2 --- 3 Acetic Acid, 30% 1 2 2 2 1 --- 2 1 2 3 Acetic Acid, 5% 1 2 2 2 1 --- 2 1 1 3 Acetic Acid, Glacial 1 3 3 3 3 --- 3 2 3 3 Acetic Acid, Hot, High Pressure 3 3 3 3 3 --- 3 3 3 3 Acetic Anhydride 2 3 3 3 2 --- 3 3 --- 3 Acetoacetic Acid 1 3 3 3 1 --- --- 2 --- 3 Acetone 1 3 3 3 3 --- 3 3 3 3 Acetone Cyanohydrin 1 3 3 3 1 --- --- 2 --- 3 Acetonitrile 1 3 3 3 1 --- --- --- --- 3 Acetophenetidine 3 2 2 2 3 --- --- --- --- 1 Acetophenone 1 3 3 3 3 --- 3 3 --- 3 Acetotoluidide 3 2 2 2 3 --- --- --- --- 1 Acetyl Acetone 1 3 3 3 3 --- 3 3 --- 3 The data and recommendations presented are based upon the best information available resulting from a combination of Victaulic's field experience, laboratory testing and recommendations supplied by prime producers of basic copolymer materials. -

EPDM & FKM Chemical Resistance Guide
EPDM & FKM Chemical Resistance Guide FIRST EDITION EPDM & FKM CHEMICAL RESISTANCE GUIDE Elastomers: Ethylene Propylene (EPDM) Fluorocarbon (FKM) Chemical Resistance Guide Ethylene Propylene (EPDM) & Fluorocarbon (FKM) 1st Edition © 2019 by IPEX. All rights reserved. No part of this book may be used or reproduced in any manner whatsoever without prior written permission. For information contact: IPEX, Marketing, 1425 North Service Road East, Oakville, Ontario, Canada, L6H 1A7 ABOUT IPEX At IPEX, we have been manufacturing non-metallic pipe and fittings since 1951. We formulate our own compounds and maintain strict quality control during production. Our products are made available for customers thanks to a network of regional stocking locations from coast-to-coast. We offer a wide variety of systems including complete lines of piping, fittings, valves and custom-fabricated items. More importantly, we are committed to meeting our customers’ needs. As a leader in the plastic piping industry, IPEX continually develops new products, modernizes manufacturing facilities and acquires innovative process technology. In addition, our staff take pride in their work, making available to customers their extensive thermoplastic knowledge and field experience. IPEX personnel are committed to improving the safety, reliability and performance of thermoplastic materials. We are involved in several standards committees and are members of and/or comply with the organizations listed on this page. For specific details about any IPEX product, contact our customer service department. INTRODUCTION Elastomers have outstanding resistance to a wide range of chemical reagents. Selecting the correct elastomer for an application will depend on the chemical resistance, temperature and mechanical properties needed. Resistance is a function both of temperatures and concentration, and there are many reagents which can be handled for limited temperature ranges and concentrations. -

List of Chemical Resistance of Materials in Contact with Liquid ANALYSETTE 22 Next & ANALYSETTE 28 Imagesizer LASER PARTICLE SIZER & PARTICLE SIZER
List of chemical resistance of materials in contact with liquid ANALYSETTE 22 NeXT & ANALYSETTE 28 ImageSizer LASER PARTICLE SIZER & PARTICLE SIZER This chart information is based on FRITSCH's knowledge and experience and are intended as a guide and not as a guarantee. We cannot assure the perfect performance of the materials as that depends on many factors as working pressure, pressure picks, fluid, ambient temperature, fluid temperature, concentra‐ tion, duration of exposure, etc… Fritsch GmbH Milling and Sizing Industriestrasse 8 D - 55743 Idar-Oberstein Telephone: +49 (0) 6784/ 70-0 Email: [email protected] Internet: www.fritsch.de Version 04/2020 Index 001 Table of contents Table of contents 1 Materials in contact with liquid.................................................... 4 2 Abbreviations................................................................................. 5 3 List of chemical resistance............................................................. 7 3.1 KEY......................................................................................... 7 3.2 Note....................................................................................... 7 3.3 Table 1.................................................................................... 7 3.4 Table 2.................................................................................. 97 3.5 Table 3................................................................................ 105 - 3 - Materials in contact with liquid 1 Materials in contact with liquid Wet dispersion -

CHEMICAL RESISTANCE CHART Chemical Resistance Glove Chart
CHEMICAL RESISTANCE CHART Chemical Resistance Glove Chart P = Poor | F = Fair | G = Good | E = Excellent | X = Not Tested Chemical Latex Nitrile 1,1,1,2-Tetrachloroethane P F P 1,1,1-Trichloroethane P P P 1,1,2-Trichloroethane P P P 1,2,4,5-Tetrachlorobenzene X E P 1,2,4-Trichlorobenzene P F P 1,2-Dichlorobenzene (o-Dichlorobenzene) P P P 1,2-Dichloroethylene P P P 1,2-Propylenoxide P P P 1,3-Dioxane F P P 1,4-Dioxane P P P 1-Nitropropane G P P 2-(Dietylamino)ethanol P E P 2-Chloroethanol X P P 2-Hydroxyethyl acrylate P P P 2-Hydroxyethyl methacrylate (HEMA) X X X 2-Nitropropane P P P 2-Nitrotoluene P P P 3-Bromopropionic acid G F X 4,4-Methylenedianiline (MDA) X X X 5-Fluorouracil X X X Acetaldehyde G P P Acetamide G P P Acetate solvent G F P Acetic acid (glacial acetic acid) 99+ % G F F Acetic acid 30% E G F Acetic acid anhydride G F P Acetone F P P Acetonitrile F F P Acetyl chloride P P P Acetylene G E F Acryl Acid P F P Acrylamide, 30-70% P E F Acrylic Acid G G P Acrylonitrile P P P Adipic acid E E X Allyl alcohol P P P Allylamine P P P Allylchloride (3-Chloroporpene) P P P Aluminium Acetate E G X Aluminium Chloride E E G Aluminium Fluoride G E F Aluminium Hydroxide P E F Aluminium Nitrate E E X Aluminium Potassium Sulfate E E F Aluminium Sulfate E E G Amine G P Ammonia anhdyrous P G G Ammonia gas (cold) E E X Ammonia gas (hot) P P X Ammonia Nitrate X F X 74 Chemical Resistance Glove Chart P = Poor | F = Fair | G = Good | E = Excellent | X = Not Tested Chemical Latex Nitrile Ammonia solution, 30% P E X Ammonium Acetate E E X Ammonium -
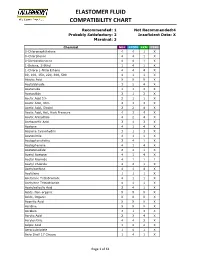
Elastomer Fluid Compatibility Chart
ELASTOMER FLUID COMPATIBILITY CHART Recommended: 1 Not Recommended:4 Probably Satisfactory: 2 Insuficient Data: X Marginal: 3 Chemical NBR EPDM FKM PTFE 0-Chloronaphthalene 4 4 1 X 0-Chlorphenol 4 4 1 X 0-Dichlorobenzene 4 4 1 X 1-Butene, 2-Ethyl 1 4 1 X 1-Chloro 1-Nitro Ethane 4 4 4 X 90, 100, 150, 220, 300, 500 4 1 1 X Abietic Acid X X X X Acetaldehyde 3 2 4 X Acetamide 1 1 3 X Acetanilide 3 1 3 X Acetic Acid 5% 2 1 1 X Acetic Acid, 30% 2 1 3 X Acetic Acid, Glacial 2 2 4 X Acetic Acid, Hot, High Pressure 4 3 4 X Acetic Anhydride 4 2 4 X Acetoacetic Acid 3 1 3 X Acetone 4 1 4 X Acetone Cyanohydrin 3 1 3 X Acetonitrile 3 1 1 X Acetophenetidine 2 4 1 X Acetophenone 4 1 4 X Acetotoluidide 2 4 1 X Acetyl Acetone 4 1 4 X Acetyl Bromide 4 1 1 1 Acetyl Chloride 4 4 1 X Acetylacetone 4 1 4 X Acetylene 1 1 1 X Acetylene Tetrabromide 4 1 1 X Acetylene Tetrachloride 4 1 1 X Acetylsalicylic Acid 2 4 1 X Acids, Non-organic X X X X Acids, Organic X X X X Aconitic Acid X X X X Acridine X X X X Acrolein 3 1 3 X Acrylic Acid 2 3 4 X Acrylonitrile 4 4 3 X Adipic Acid 1 2 2 X Aero Lubriplate 1 4 1 X Aero Shell 17 Grease 1 4 1 X Page 1 of 61 ELASTOMER FLUID COMPATIBILITY CHART Recommended: 1 Not Recommended:4 Probably Satisfactory: 2 Insuficient Data: X Marginal: 3 Chemical NBR EPDM FKM PTFE Aero Shell 1AC Grease 1 4 1 X Aero Shell 750 2 4 1 X Aero Shell 7A Grease 1 4 1 X Aerosafe 2300 4 1 4 X Aerosafe 2300W 4 1 4 X Aerozene 50 (50% Hydrazine 50% UDMH) 3 1 4 X Air, 1 1 1 X Air, 200 - 300° F 3 2 1 X Air, 300 - 400° F 4 4 1 X Air, 400 - 500° F 4 4 3 X Air, -
Aminoxyl Radicals) in Biochemistry: Past, Present and Future of Spin Label and Probe Method
Chapter 1 History of the Use of Nitroxides (Aminoxyl Radicals) in Biochemistry: Past, Present and Future of Spin Label and Probe Method Lawrence J. Berliner Additional information is available at the end of the chapter http://dx.doi.org/10.5772/39115 1. Introduction The perspective of this chapter is very much historical. The author was fortunate enough to have begun his graduate studies at the very inception of the technique of spin labeling. Mind you, the topic of this book is nitroxides a.k.a. aminoxyl radicals which in fact preceded the spin labeling method and its inception. Hence the chapter will cover a history of the synthetic developments with nitroxides, the history of the development of spin labels, and the use of nitroxides and will provide an overview to the future of its applications. The intent is to cover the very beginning and then discuss some of the key areas (always dominated by synthetic organic chemistry) that allowed this technique to blossom more and more. Needless to say, while the definition of spin labeling is the incorporation of a stable, free radical into a macromolecular system of choice, we have yet to find anything (aside from the trityl radicals) that will fulfill this purpose. And the trityl radicals, which may be covered briefly in this chapter, give no dynamic or structural information whatsoever as they yield a single narrow line spectrum. The author feels confident to discuss these areas since he was deeply involved in the synthetic organic chemistry of these compounds, frequently repeating the syntheses of the basic starting compounds that were either commercially unavailable or quite expensive at the time. -

Chestertonrotating.Com
SEAL RECOMMENDATIONS BY FLUID www.chestertonrotating.com Form. EN23874 03/15 Seal Recommendations by Fluid CONCENTRATION AND PIPING PLANS Code - * : Substance currently does not have The recommended piping plan or choice of a CAS Number listed; therefore, substance TEMPERATURE RANGES has not been classified accordingly. Concentration: Listed as a percentage of plans to be utilized to enhance seal life and the pure component. The diluent for all non performance based on the API Auxiliary Piping No Code: Substance is not classified under the 100% entries is water unless otherwise noted. Systems for mechanical seals. There is no parameters specified in the regulations. It does <SP indicates the seal recommendation is valid piping plan designated for applications which not necessarily mean that there are no hazards for any concentration less than the solubility point typically do not require one. Where more than associated with the substance. of the substance in water. one plan is listed: Temperature: Temperature or range listed in Plan 1, Plan 2 (separated by a comma) MATERIALS OF CONSTRUCTION - degrees Celsius (ºC) and Fahrenheit (ºF) for which Use the most appropriate plan for your PERFORMANCE CAPABILITIES the seal recommendation is applicable at the application. Materials of construction are grouped by specified concentration. Plan 1/Plan 2 (separated by a slash) metallurgy, faces, and secondary seals. Use both plans in conjunction for Performance capabilities of the seal materials are CAS the application. rated A = Acceptable and B = Alternate Choice. CAS numbers are internationally recognized These ratings are based on existing published and are a resource for scientists, industry, HAZARD INFORMATION data (references), laboratory tests, and informed and regulatory bodies. -

Nsf/Ansi 61 - 2016
NSF International Standard / American National Standard NSF/ANSI 61 - 2016 Drinking Water System Components - Health Effects NSF International, an independent, not- for-profit, non-governmental organization, is dedicated to being the leading global provider of public health and safety-based risk management solutions while serving the interests of all stakeholders. Not for Distribution or SaleThis Standard is subject to revision. Contact NSF to confirm this revision is current. Users of this Standard may request clarifications and interpretations, or propose revisions by contacting: Chair, Joint Committee on Drinking Water Additives c/o NSF International 789 North Dixboro Road, P.O. Box 130140 Ann Arbor, Michigan 48113-0140 USA Phone: (734) 769-8010 Telex: 753215 NSF INTL FAX: (734) 769-0109 E-mail: [email protected] Web: http://www.nsf.org NSF/ANSI 61 – 2016 International Standard/ American National Standard for Drinking Water Additives ― Drinking water system components ― Health effects Not for Distribution or Sale Standard Developer NSF International NSF International Board of Directors Designated an ANSI Standard January 05, 2016 American National Standards Institute i Prepared by The NSF Joint Committee on Drinking Water Additives Recommended for Adoption by The NSF Council of Public Health Consultants Adopted by The NSF Board of Directors June 1988 Revised October 1988 Revised November 1999 Addendum October 2007 Revised May 1990 Revised September 2000 Revised December 2008 Revised May 1991 Revised February 2001 Revised August 2009 Revised -

A Thesis Entitled * Studies in Nitroxide Radical Chemistry
A THESIS ENTITLED * STUDIES IN NITROXIDE RADICAL CHEMISTRY * SUBMITTED TO THE UNIVERSITY OF GLASGOW FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN THE FACULTY OF SCIENCE BY C. THOMSON B.Sc. CHEMISTRY DEPARTMENT SEPTEMBER, 1971 ProQuest Number: 11011976 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 11011976 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 4 8 1 0 6 - 1346 Jeanette, ACKNOWLEDGEMENTS. The author wishes to express his gratitude to Dr. J. S. Roberts for his constant help, advice, encouragement and friendship during the preparation of this work. I should also like to thank the technical staff of this department for their able assistance, Dr. A. L. Porte for obtaining the e.p.r. spectra and Drs. G. C. Wood and.P. M. Scop for running the ORD and CD spectra. A maintenance award from the Carnegie Trust for the Universities of Scotland is gratefully acknowledged. Finally I should like to thank my wife and my parents for their encouragement and for their patience in typing the manuscript. SUMMARY A suitable synthetic route to trimethyldecahydroquinoline nitroxide radicals is described. -

Dupont™ Kalrez® Chemical Resistance and Fluid Compatability, Including All Chemicals Under the Clean Air
DuPont™ Kalrez® Chemical Resistance and Fluid Compatibility, Including All Chemicals Under the Clean Air Act Technical Information—Rev. 4, July 2010 DuPont™ Kalrez® perfluoroelastomer parts combine the elastomeric properties of fluoroelastomers with the chemical resistance of DuPont™ Teflon® fluoropolymer resins. Due to its unique properties, Kalrez® parts should be considered for service in all applications and environments where dependable, long-term service is desired, as well as in hot or aggressive environments that are beyond the service ability of common elastomers. This guide is intended to provide assistance in determining the suitability of seven commercially available elastomers—nitrile (NBR), ethylene propylene (EPDM), silicone (VMQ), fluorosilicone (FVMQ), vinylidene fluoride-based fluoroelastomer (FKM), polysulfides (T), and Kalrez® perfluoroelastomer parts—for service in over 1,600 chemicals and fluids. The criteria used for these ratings included volume swell resistance based on laboratory immersion testing, laboratory aging tests, actual field experience, and informed judgments based on experience in similar chemical groups. The ratings for the six common elastomers are based on published literature and are offered for general comparative purposes only—we cannot guarantee their accuracy nor assume responsibility for their use. Thermal Stability The ratings for these six common elastomers may be overly optimistic for elevated temperature and/or high concentration applications because many are based on ambient temperature testing. Suitability of these elastomers for service at elevated temperatures rapidly diminishes because higher temperatures increase the effects of chemicals on the base polymer as well as the cross-link systems. Serviceability is further limited by the upper service temperature limit of each polymer. -

105 Cmr: Department of Public Health
105 CMR: DEPARTMENT OF PUBLIC HEALTH 105 CMR 670.000: "RIGHT TO KNOW" Section 670.001: Purpose 670.005: Definitions 670.010: The Massachusetts Substance List 670.020: Trade Secrets 670.025: Physician's Access to Material Safety Data Sheets Appendix A 670.001: Purpose The purpose of 105 CMR 670.000 is to protect the public health by providing and encouraging the greatest possible transmission of health and safety information concerning toxic and hazardous substances. 670.005: Definitions As used in 105 CMR 670.000 the following words and phrases have the following meanings: Carcinogen means: (1) any substance or combination of substances which causes an increased incidence of benign and/or malignant neoplasms in one or more species; or, (2) any substance or combination of substances which substantially decreases the latency period between exposure and onset of a neoplasm in one or more species; or, (3) any substance or combination of substances which is metabolized into one or more substances that causes an increased incidence of neoplasms or decreases the latency period between exposure and onset of neoplasms in one or more species. CAS Number means the identification number assigned by the Chemical Abstracts Service to a specific chemical substance. Chemical Name means the scientific designation of a substance in accordance with the nomenclature system developed by the International Union of Pure and Applied Chemistry, or the system developed by the Chemical Abstracts Service. Commissioner means the Commissioner of Public Health. Common Name means any designation or identification such as a code name, code number, trade name, or brand name used to identify a substance other than by its chemical name. -

Hazardous Chemicals Handbook
Hazardous Chemicals Handbook Hazardous Chemicals Handbook Second edition Phillip Carson PhD MSc AMCT CChem FRSC FIOSH Head of Science Support Services, Unilever Research Laboratory, Port Sunlight, UK Clive Mumford BSc PhD DSc CEng MIChemE Consultant Chemical Engineer Oxford Amsterdam Boston London New York Paris San Diego San Francisco Singapore Sydney Tokyo Butterworth-Heinemann An imprint of Elsevier Science Linacre House, Jordan Hill, Oxford OX2 8DP 225 Wildwood Avenue, Woburn, MA 01801-2041 First published 1994 Second edition 2002 Copyright © 1994, 2002, Phillip Carson, Clive Mumford. All rights reserved The right of Phillip Carson and Clive Mumford to be identified as the authors of this work has been asserted in accordance with the Copyright, Designs and Patents Act 1988 No part of this publication may be reproduced in any material form (including photocopying or storing in any medium by electronic means and whether or not transiently or incidentally to some other use of this publication) without the written permission of the copyright holder except in accordance with the provisions of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London, England W1T 4LP. Applications for the copyright holder’s written permission to reproduce any part of this publication should be addressed to the publishers British Library Cataloguing in Publication Data A catalogue record for this book is available from the British Library Library of Congress Cataloguing