Revisiting the Taxonomic Synonyms and Populations of Saccharomyces Cerevisiae—Phylogeny, Phenotypes, Ecology and Domestication
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Brewing Yeast – Theory and Practice
Brewing yeast – theory and practice Chris Boulton Topics • What is brewing yeast? • Yeast properties, fermentation and beer flavour • Sources of yeast • Measuring yeast concentration The nature of yeast • Yeast are unicellular fungi • Characteristics of fungi: • Complex cells with internal organelles • Similar to plants but non-photosynthetic • Cannot utilise sun as source of energy so rely on chemicals for growth and energy Classification of yeast Kingdom Fungi Moulds Yeast Mushrooms / toadstools Genus > 500 yeast genera (Means “Sugar fungus”) Saccharomyces Species S. cerevisiae S. pastorianus (ale yeast) (lager yeast) Strains Many thousands! Biology of ale and lager yeasts • Two types indistinguishable by eye • Domesticated by man and not found in wild • Ale yeasts – Saccharomyces cerevisiae • Much older (millions of years) than lager strains in evolutionary terms • Lot of diversity in different strains • Lager strains – Saccharomyces pastorianus (previously S. carlsbergensis) • Comparatively young (probably < 500 years) • Hybrid strains of S. cerevisiae and wild yeast (S. bayanus) • Not a lot of diversity Characteristics of ale and lager yeasts Ale Lager • Often form top crops • Usually form bottom crops • Ferment at higher temperature o • Ferment well at low temperatures (18 - 22 C) (5 – 10oC) • Quicker fermentations (few days) • Slower fermentations (1 – 3 weeks) • Can grow up to 37oC • Cannot grow above 34oC • Fine well in beer • Do not fine well in beer • Cannot use sugar melibiose • Can use sugar melibiose Growth of yeast cells via budding + + + + Yeast cells • Each cell is ca 5 – 10 microns in diameter (1 micron = 1 millionth of a metre) • Cells multiply by budding a b c d h g f e Yeast and ageing - cells can only bud a certain number of times before death occurs. -

Multiple Genetic Origins of Saccharomyces Cerevisiae in Bakery
Multiple genetic origins of Saccharomyces cerevisiae in bakery Frédéric Bigey, Diego Segond, Lucie Huyghe, Nicolas Agier, Aurélie Bourgais, Anne Friedrich, Delphine Sicard To cite this version: Frédéric Bigey, Diego Segond, Lucie Huyghe, Nicolas Agier, Aurélie Bourgais, et al.. Multiple genetic origins of Saccharomyces cerevisiae in bakery. Comparative genomics of eukaryotic microbes: genomes in flux, and flux between genomes, Oct 2019, Sant Feliu de Guixol, Spain. hal-02950887 HAL Id: hal-02950887 https://hal.archives-ouvertes.fr/hal-02950887 Submitted on 28 Sep 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. genetic diversity of229 propagating a natural sourdough, which is a of mixed naturally fermented domestication of wide diversity of fermented products like wine, Saccharomyces cerevisiae sake, beer, cocoa and bread. While bread is of cultural and historical importance, the highly diverse environments like human, wine, sake, fruits, tree, soil... fruits, tree, human, wine, sake, like highly diverse environments clade on the 1002 genomes whichtree, also includes beer strains. Sourdough strains mosaic are and genetically are to related strains from strains. Selection for strains commercial has maintained autotetraploid. strains mostly to Commercial are related strains of origin the Mixed The origin of bakery strains is polyphyletic. -

Relation Between the Recipe of Yeast Dough Dishes and Their Glycaemic Indices and Loads
foods Article Relation between the Recipe of Yeast Dough Dishes and Their Glycaemic Indices and Loads Ewa Raczkowska * , Karolina Ło´zna , Maciej Bienkiewicz, Karolina Jurczok and Monika Bronkowska Department of Human Nutrition, Faculty of Biotechnology and Food Sciences, Wrocław University of Environmental and Life Sciences, 51-630 Wroclaw, Poland * Correspondence: [email protected]; Tel.: +48-71-320-7726 Received: 23 July 2019; Accepted: 30 August 2019; Published: 1 September 2019 Abstract: The aim of the study was to evaluate the glycaemic indices (GI) and glycaemic loads (GL) of four food dishes made from yeast dough (steamed dumplings served with yoghurt, apple pancakes sprinkled with sugar powder, rolls with cheese and waffles with sugar powder), based on their traditional and modified recipes. Modification of the yeast dough recipe consisted of replacing wheat flour (type 500) with whole-wheat flour (type 2000). Energy value and the composition of basic nutrients were assessed for every tested dish. The study was conducted on 50 people with an average age of 21.7 1.1 years, and an average body mass index of 21.2 2.0 kg/m2. The GI of the analysed ± ± food products depended on the total carbohydrate content, dietary fibre content, water content, and energy value. Modification of yeast food products by replacing wheat flour (type 500) with whole-wheat flour (type 2000) contributed to the reduction of their GI and GL values, respectively. Keywords: glycaemic index; glycaemic load; yeast dough 1. Introduction In connection with the growing number of lifestyle diseases, consumers pay increasing attention to food, not only to food that have a better taste but also to food that help maintain good health. -

CONTROL NUMBER 27. W ' Cu-.1.SSJT:ON(€S)
m ,. CONTROL NUMBER 27.SU' W' cu-.1.SSJT:ON(€S) BIBLIOGRAPHIC DATA SHEET AL-3L(-UM0F00UIEC-T00J: 9 i IE AN) SUBTIT"LE (.40) - Airicult !ral development : present and potential role of edible wild plants. Part I Central .nd So,ith :Amnerica a: ( the Caribbean 4af.RSON \1. AUrI'hORS (100) 'rivetti, L. E. 5. CORPORATE At-TH1ORS (A01) Calif. Uiliv., 1)' is. 6. nOX:U1E.N7 DATE 0)0' 1. NUMBER 3F PAGES j12*1) RLACtNUMER ([Wr I icio 8 2 p. J631.54.,G872 9. REI F.RENCE OR(GA,-?A- ION (1SO) C,I i f. -- :v., 10. SIt J'PPNIENTA.RY NOTES (500) (Part II, Suh-Sanarai Africa PN-AAJ-639; Part III, India, East Asi.., Southeast Asi cearnia PAJ6)) 11. NBS'RACT (950) 12. DESCRIPTORS (920) I. PROJECT NUMBER (I) Agricultural development Diets Plants Wild plants 14. CONTRATr NO.(I. ) i.CONTRACT Food supply Caribbean TYE(140) Central America AID/OTR-147-80-87 South America 16. TYPE OF DOCI M.NT ic: AED 590-7 (10-79) IL AGRICULTURAL DEVELOP.MMT: PRESENT AND POTENI.AL ROLE OF EDIBLE WILD PLANTS PART I CENTRAL AND SOUTH AMERICA AND THE CARIBBEAN November 1980 REPORT TO THE DEPARTMENT OF STATE AGENCY FOR INTERNATIONAL DEVELOPMENT Alb/mr-lq - 6 _ 7- AGRICULTURAL DEVELOPMENT: PRESENT AND POTENTIAL ROLE OF EDIBLE WILD PLANTS PART 1 CENTRAL AND SOUTH AMERICA AND THE CARIBBEAN by Louis Evan Grivetti Departments of Nutrition and Geography University of California Davis, California 95616 With the Research Assistance of: Christina J. Frentzel Karen E. Ginsberg Kristine L. -

Sensory Evaluation and Staling of Bread Produced by Mixed Starter of Saccharomyces Cerevisiae and L.Plantarum
J. Food and Dairy Sci., Mansoura Univ., Vol. 5 (4): 221 - 233, 2014 Sensory evaluation and staling of bread produced by mixed starter of saccharomyces cerevisiae and l.plantarum Swelim, M. A. / Fardous M. Bassouny / S. A. El-Sayed / Nahla M. M. Hassan / Manal S. Ibrahim. * Botany Department, Faculty of science, Banha University. ** Agricultural Microbiology Department, Agricultural Research Center, Giza, Egypt. ***Food Technology Research Institute, Agricultural Research Center, Giza, Egypt. ABSTRACT Impact of processed conditions and type of starter cultivars on characteristics of wheat dough bread by using mixed starter cultivars of Sacch. cerevisiae and L. plantarum was estimated. L. plantarum seemed to be more effective in combination with Sacch. cerevisiae on the dough volume. Dough produced by starter containing L. plantarum characterized with lower PH and higher total titratable acidity (TTA) and moisture content, in comparison with control treatment. Highly significant differences in aroma and crumb texture were recorded with bread produced by starter culture containing L. plantarum. The obtained results also revealed significant differences in bread firmness which reflected the staling and its rate among the tested treatments after 1, 3, 5 and 6 days during storage time at room temperature. Data also confirmed a processing technique using L. planturium mixed with Sacch.cerevisia to enhance organoleptic properties of produced bread. INTRODUCTION Cereal fermentation is one of the oldest biotechnological processes, dating back to ancient Egypt, where both beer and bread were produced by using yeasts and lactic acid bacteria (LAB), Clarke and Arendt, (2005). Starters composed of specific individual LAB, or mixed with yeasts, became available a few years ago allowing the production of a full sourdough in one- stage process. -

AP-42, CH 9.13.4: Yeast Production
9.13.4 Yeast Production 9.13.4.1 General1 Baker’s yeast is currently manufactured in the United States at 13 plants owned by 6 major companies. Two main types of baker’s yeast are produced, compressed (cream) yeast and dry yeast. The total U. S. production of baker’s yeast in 1989 was 223,500 megagrams (Mg) (245,000 tons). Of the total production, approximately 85 percent of the yeast is compressed (cream) yeast, and the remaining 15 percent is dry yeast. Compressed yeast is sold mainly to wholesale bakeries, and dry yeast is sold mainly to consumers for home baking needs. Compressed and dry yeasts are produced in a similar manner, but dry yeasts are developed from a different yeast strain and are dried after processing. Two types of dry yeast are produced, active dry yeast (ADY) and instant dry yeast (IDY). Instant dry yeast is produced from a faster-reacting yeast strain than that used for ADY. The main difference between ADY and IDY is that ADY has to be dissolved in warm water before usage, but IDY does not. 9.13.4.2 Process Description1 Figure 9.13.4-1 is a process flow diagram for the production of baker’s yeast. The first stage of yeast production consists of growing the yeast from the pure yeast culture in a series of fermentation vessels. The yeast is recovered from the final fermentor by using centrifugal action to concentrate the yeast solids. The yeast solids are subsequently filtered by a filter press or a rotary vacuum filter to concentrate the yeast further. -
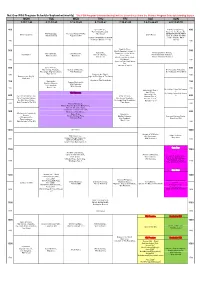
Nat Geo Wild Program Schedule September(Weekly)
Nat Geo Wild Program Schedule September(weekly) *The FOX Program Schedule(weekly) will be discontinued from the October Program Schedule(starting Septem MON TUE WED THU FRI SAT SUN 3.10.17.24 4.11.18.25 5.12.19.26 6.13.20.27 7.14.21.28 1.8.15.22.29 2.9.16.23.30 400 Game Of Lions、 And Man Created Dog、 400 Man Among Cheetahs、 Cesar To The Rescue III、 Wild Mississippi、 America's National Parks、 Tiger Queen SEARCH FOR THE FIRST World's Deadliest Shark Men 2 Wild Scotland II Expedition Wild DOG: A QUEST TO FIND [20th]NO TRANSMISSION DUE OUR ORIGINAL BEST TO MAINTENANCE(~7:00) FRIEND 430 430 Dead Or Alive、 500 World's Deadliest Animals 3、 500 Superpride、 World's Deadliest Animals、 Wild Cambodia、 Safari Brothers、 Symphony For Our World、 Shark Men 2 Big Cat Odyssey、 World's Deadliest Animals 2、 Wild Colombia Mygrations Shark Eden、 Man V. Lion Life On The Barrier Reef、 World's Deadliest Animals 3 530 Wild Hawaii、 530 Secrets of the Giant Manta Ray、 Moment of Impact 600 Game Of Lions、 600 Man Among Cheetahs、 Secrets of Wild India、 Dr. K's Exotic Animal ER、 The Jungle King Compilation、 Wild Mississippi Dr. K's Exotic Animal ER 2 Tiger Queen 630 Snakes in the City II、 630 Snakes in the City II、 Light At The Edge of The World Earth Live 2、 Superpride、 Kingdom of The Blue Whale 700 Amazon Underworld、 700 Big Cat Odyssey、 Wild Cambodia、 Lion Gangland、 Wild Colombia Man V. Lion 730 730 Dr. -

Table S1 V4.Xlsx
Table S1. Strains and genomes used in this study and relevant information pertaining to them. Clime Zone 2 Regions BIO1 Strain Other designations Original species designation (correspondent type strain) Substrate Geographic location (wild /feral Phylogeny Clade Heterozygosity1 AQY13 AQY23 RTM14 STA16 MEL7 GENOME DATA REFERENCE BIO65 pop.) ABC CBS 1175 Saccharomyces cerevisiae var. orsati Wine Switzerland WINE - Main 1 3522 15/15 NF A881del NF 11bp del x x x x ERP014555 Peter et al. 2018 CBS 1194 Saccharomyces ellipsoideus var. thermophilus Wine Unknown WINE - Main 1 1597 NF A881del NF 11bp del x x x x ERP014555 Peter et al. 2018 CBS 1479 Saccharomyces ellipsoideus var. montibensis Wine Unknown WINE - Main 1 1083 NF A881del NF 11bp del x x x x ERP014555 Peter et al. 2018 CBS 1192 Saccharomyces ellipsoideus var. alpinus Wine Unknown WINE - Main 1 1183 15/15 18/19 NF A881del NF 11bp del x x x x ERP014555 Peter et al. 2018 CBS 423 PYCC 2607 Saccharomyces chodati Wine Switzerland WINE - Main 1 4813 15/15 18/19 NF A881del NF 11bp del x x x x ERP014555 Peter et al. 2018 CBS 436 PYCC 8195 Saccharomyces yedo Sake moto Japan WINE - Main 1 n.d. 5/5 18/19 NF A881del NF 11bp del x x x x PRJEB36095 This study CBS 457 Saccharomyces ellipsoideus var. umbra Grape must Italy WINE - Main 1 5501 18/19 NF A881del NF 11bp del x x x x ERP014555 Peter et al. 2018 CBS 459 Saccharomyces italicus Grape must Italy WINE - Main 1 1095 NF A881del NF 11bp del x x x x ERP014555 Peter et al. -

Tersteege 1..10
ORE Open Research Exeter TITLE Estimating the global conservation status of more than 15,000 Amazonian tree species AUTHORS ter Steege, H; Pitman, Nigel C.A.; Killeen, TJ; et al. JOURNAL Science Advances DEPOSITED IN ORE 12 January 2016 This version available at http://hdl.handle.net/10871/19212 COPYRIGHT AND REUSE Open Research Exeter makes this work available in accordance with publisher policies. A NOTE ON VERSIONS The version presented here may differ from the published version. If citing, you are advised to consult the published version for pagination, volume/issue and date of publication RESEARCH ARTICLE ECOLOGY 2015 © The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. Distributed Estimating the global conservation status of under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC). more than 15,000 Amazonian tree species 10.1126/sciadv.1500936 Hans ter Steege,1,2* Nigel C. A. Pitman,3,4 Timothy J. Killeen,5 William F. Laurance,6 Carlos A. Peres,7 Juan Ernesto Guevara,8,9 Rafael P. Salomão,10 Carolina V. Castilho,11 Iêda Leão Amaral,12 FranciscaDioníziadeAlmeidaMatos,12 Luiz de Souza Coelho,12 William E. Magnusson,13 Oliver L. Phillips,14 Diogenes de Andrade Lima Filho,12 Marcelo de Jesus Veiga Carim,15 Mariana Victória Irume,12 Maria Pires Martins,12 Jean-François Molino,16 Daniel Sabatier,16 Florian Wittmann,17 Dairon Cárdenas López,18 José Renan da Silva Guimarães,15 Abel Monteagudo Mendoza,19 Percy Núñez Vargas,20 Angelo Gilberto Manzatto,21 Neidiane Farias Costa Reis,22 John Terborgh,4 Katia Regina Casula,22 Juan Carlos Montero,12,23 Ted R. -

14419802.Pdf
View metadata, citation and similar papers at core.ac.uk brought to you by CORE REVIEW ARTICLE 277 Potential Application of Yeast β-Glucans in Food Industry Vesna ZECHNER-KRPAN 1 Vlatka PETRAVIĆ-TOMINAC 1 ( ) Ines PANJKOTA-KRBAVČIĆ 2 Slobodan GRBA 3 Katarina BERKOVIĆ 4 Summary Diff erent β-glucans are found in a variety of natural sources such as bacteria, yeast, algae, mushrooms, barley and oat. Th ey have potential use in medicine and pharmacy, food, cosmetic and chemical industries, in veterinary medicine and feed production. Th e use of diff erent β-glucans in food industry and their main characteristics important for food production are described in this paper. Th is review focuses on benefi cial properties and application of β-glucans isolated from diff erent yeasts, especially those that are considered as waste from brewing industry. Spent brewer’s yeast, a by-product of beer production, could be used as a raw-material for isolation of β-glucan. In spite of the fact that large quantities of brewer’s yeast are used as a feedstuff , certain quantities are still treated as a liquid waste. β-Glucan is one of the compounds that can achieve a greater commercial value than the brewer’s yeast itself and maximize the total profi tability of the brewing process. β-Glucan isolated from spent brewer’s yeast possesses properties that are benefi cial for food production. Th erefore, the use of spent brewer’s yeast for isolation of β-glucan intended for food industry would represent a payable technological and economical choice for breweries. -

Tersteege 1..10
RESEARCH ARTICLE ECOLOGY 2015 © The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. Distributed Estimating the global conservation status of under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC). more than 15,000 Amazonian tree species 10.1126/sciadv.1500936 Hans ter Steege,1,2* Nigel C. A. Pitman,3,4 Timothy J. Killeen,5 William F. Laurance,6 Carlos A. Peres,7 Juan Ernesto Guevara,8,9 Rafael P. Salomão,10 Carolina V. Castilho,11 Iêda Leão Amaral,12 FranciscaDioníziadeAlmeidaMatos,12 Luiz de Souza Coelho,12 William E. Magnusson,13 Oliver L. Phillips,14 Diogenes de Andrade Lima Filho,12 Marcelo de Jesus Veiga Carim,15 Mariana Victória Irume,12 Maria Pires Martins,12 Jean-François Molino,16 Daniel Sabatier,16 Florian Wittmann,17 Dairon Cárdenas López,18 José Renan da Silva Guimarães,15 Abel Monteagudo Mendoza,19 Percy Núñez Vargas,20 Angelo Gilberto Manzatto,21 Neidiane Farias Costa Reis,22 John Terborgh,4 Katia Regina Casula,22 Juan Carlos Montero,12,23 Ted R. Feldpausch,14,24 Euridice N. Honorio Coronado,14,25 Alvaro Javier Duque Montoya,26 Charles Eugene Zartman,12 Bonifacio Mostacedo,27 Rodolfo Vasquez,19 Rafael L. Assis,28 Marcelo Brilhante Medeiros,29 Downloaded from Marcelo Fragomeni Simon,29 Ana Andrade,30 José Luís Camargo,30 Susan G. W. Laurance,6 Henrique Eduardo Mendonça Nascimento,12 Beatriz S. Marimon,31 Ben-Hur Marimon Jr.,31 Flávia Costa,13 Natalia Targhetta,28 Ima Célia Guimarães Vieira,10 Roel Brienen,14 Hernán Castellanos,32 Joost F. Duivenvoorden,33 Hugo F. -

Impact of Wort Amino Acids on Beer Flavour: a Review
fermentation Review Impact of Wort Amino Acids on Beer Flavour: A Review Inês M. Ferreira and Luís F. Guido * LAQV/REQUIMTE, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre, 687, 4169-007 Porto, Portugal; ines.fi[email protected] * Correspondence: [email protected]; Tel.: +351-220-402-644 Received: 3 March 2018; Accepted: 25 March 2018; Published: 28 March 2018 Abstract: The process by which beer is brewed has not changed significantly since its discovery thousands of years ago. Grain is malted, dried, crushed and mixed with hot water to produce wort. Yeast is added to the sweet, viscous wort, after which fermentation occurs. The biochemical events that occur during fermentation reflect the genotype of the yeast strain used, and its phenotypic expression is influenced by the composition of the wort and the conditions established in the fermenting vessel. Although wort is complex and not completely characterized, its content in amino acids indubitably affects the production of some minor metabolic products of fermentation which contribute to the flavour of beer. These metabolic products include higher alcohols, esters, carbonyls and sulfur-containing compounds. The formation of these products is comprehensively reviewed in this paper. Furthermore, the role of amino acids in the beer flavour, in particular their relationships with flavour active compounds, is discussed in light of recent data. Keywords: amino acids; beer; flavour; higher alcohols; esters; Vicinal Diketones (VDK); sulfur compounds 1. Introduction The process by which beer has been brewed has not changed significantly since its discovery over 2000 years ago. Although industrial equipment is used for modern commercial brewing, the principles are the same.