Heterologous Expression and Characterization of Bacterial 2-C-Methyl-D-Erythritol-4- Phosphate Pathway in Saccharomyces Cerevisiae
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Adaptive Laboratory Evolution Enhances Methanol Tolerance and Conversion in Engineered Corynebacterium Glutamicum
ARTICLE https://doi.org/10.1038/s42003-020-0954-9 OPEN Adaptive laboratory evolution enhances methanol tolerance and conversion in engineered Corynebacterium glutamicum Yu Wang 1, Liwen Fan1,2, Philibert Tuyishime1, Jiao Liu1, Kun Zhang1,3, Ning Gao1,3, Zhihui Zhang1,3, ✉ ✉ 1234567890():,; Xiaomeng Ni1, Jinhui Feng1, Qianqian Yuan1, Hongwu Ma1, Ping Zheng1,2,3 , Jibin Sun1,3 & Yanhe Ma1 Synthetic methylotrophy has recently been intensively studied to achieve methanol-based biomanufacturing of fuels and chemicals. However, attempts to engineer platform micro- organisms to utilize methanol mainly focus on enzyme and pathway engineering. Herein, we enhanced methanol bioconversion of synthetic methylotrophs by improving cellular tolerance to methanol. A previously engineered methanol-dependent Corynebacterium glutamicum is subjected to adaptive laboratory evolution with elevated methanol content. Unexpectedly, the evolved strain not only tolerates higher concentrations of methanol but also shows improved growth and methanol utilization. Transcriptome analysis suggests increased methanol con- centrations rebalance methylotrophic metabolism by down-regulating glycolysis and up- regulating amino acid biosynthesis, oxidative phosphorylation, ribosome biosynthesis, and parts of TCA cycle. Mutations in the O-acetyl-L-homoserine sulfhydrylase Cgl0653 catalyzing formation of L-methionine analog from methanol and methanol-induced membrane-bound transporter Cgl0833 are proven crucial for methanol tolerance. This study demonstrates the importance of -

Contig Protein Description Symbol Anterior Posterior Ratio
Table S2. List of proteins detected in anterior and posterior intestine pooled samples. Data on protein expression are mean ± SEM of 4 pools fed the experimental diets. The number of the contig in the Sea Bream Database (http://nutrigroup-iats.org/seabreamdb) is indicated. Contig Protein Description Symbol Anterior Posterior Ratio Ant/Pos C2_6629 1,4-alpha-glucan-branching enzyme GBE1 0.88±0.1 0.91±0.03 0.98 C2_4764 116 kDa U5 small nuclear ribonucleoprotein component EFTUD2 0.74±0.09 0.71±0.05 1.03 C2_299 14-3-3 protein beta/alpha-1 YWHAB 1.45±0.23 2.18±0.09 0.67 C2_268 14-3-3 protein epsilon YWHAE 1.28±0.2 2.01±0.13 0.63 C2_2474 14-3-3 protein gamma-1 YWHAG 1.8±0.41 2.72±0.09 0.66 C2_1017 14-3-3 protein zeta YWHAZ 1.33±0.14 4.41±0.38 0.30 C2_34474 14-3-3-like protein 2 YWHAQ 1.3±0.11 1.85±0.13 0.70 C2_4902 17-beta-hydroxysteroid dehydrogenase 14 HSD17B14 0.93±0.05 2.33±0.09 0.40 C2_3100 1-acylglycerol-3-phosphate O-acyltransferase ABHD5 ABHD5 0.85±0.07 0.78±0.13 1.10 C2_15440 1-phosphatidylinositol phosphodiesterase PLCD1 0.65±0.12 0.4±0.06 1.65 C2_12986 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase delta-1 PLCD1 0.76±0.08 1.15±0.16 0.66 C2_4412 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2 PLCG2 1.13±0.08 2.08±0.27 0.54 C2_3170 2,4-dienoyl-CoA reductase, mitochondrial DECR1 1.16±0.1 0.83±0.03 1.39 C2_1520 26S protease regulatory subunit 10B PSMC6 1.37±0.21 1.43±0.04 0.96 C2_4264 26S protease regulatory subunit 4 PSMC1 1.2±0.2 1.78±0.08 0.68 C2_1666 26S protease regulatory subunit 6A PSMC3 1.44±0.24 1.61±0.08 -

Biosynthèse De Nouveaux Dérivés De L'alpha-Bisabolol Par Une Approche
THÈSE En vue de l’obtention du DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE Délivré par l'Institut National des Sciences Appliquées de Toulouse Présentée et soutenue par Arthur SARRADE-LOUCHEUR Le 30 juin 2020 Biosynthèse de nouveaux dérivés de l'α-bisabolol par une approche de biologie synthèse Ecole doctorale : SEVAB - Sciences Ecologiques, Vétérinaires, Agronomiques et Bioingenieries Spécialité : Ingénieries microbienne et enzymatique Unité de recherche : TBI - Toulouse Biotechnology Institute, Bio & Chemical Engineering Thèse dirigée par Gilles TRUAN et Magali REMAUD-SIMEON Jury Mme Véronique DE BERARDINIS, Rapporteure, Chercheur CEA M. Jean-Etienne BASSARD, Rapporteur, Chargé de Recherche, CNRS Mme Danièle WERCK-REICHHART, Examinatrice, Directeur de Recherche Emérite, CNRS M. Gilles TRUAN, Directeur de thèse, Directeur de Recherche, CNRS Mme Magali REMAUD-SIMEON, Co-directrice de thèse, Professeur des Universités, INSA Mme Fayza DABOUSSI, Présidente, Directeur de Recherche INRAE 2 Author: Arthur Sarrade-Loucheur Year: 2020 Biosynthesis of new α-bisabolol derivatives through a synthetic biology approach The rise of synthetic biology now enables the production of new to nature molecules. In the frame of this thesis we focused on the diversification of the (+)-epi-α-bisabolol scaffold. This molecule coming from the plant Lippia dulcis belongs to the vast family of sesquiterpenes. While sesquiterpenes possess diverse biological activities, (+)-epi-α- bisabolol is the precursor of hernandulcin, an intense sweetener. However, the last oxidative step(s) of the hernandulcin biosynthetic pathway remain elusive. Rather than seeking the native oxidase responsible for hernandulcin synthesis among L. dulcis enzymes we turned our choice towards oxidative enzymes known to be promiscuous and that could functionalize (+)-epi-α-bisabolol in order to i) generate diversity from (+)-epi-α-bisabolol; ii) hopefully identify an oxidative enzyme catalyzing hernandulcin synthesis. -

Structure of L-Rhamnose Isomerase in Complex with L
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Open Bio 3 (2013) 35–40 journal homepage: www.elsevier.com/locate/febsopenbio Structure of l-rhamnose isomerase in complex with l-rhamnopyranose demonstrates the sugar-ring opening mechanism and the role of a substrate sub-binding site Hiromi Yoshidaa, Akihide Yoshiharab, Misa Teraokaa, Satoshi Yamashitaa, Ken Izumorib, Shigehiro Kamitoria,* aLife Science Research Center and Faculty of Medicine, Kagawa University, 1750-1 Ikenobe, Miki-cho, Kita-gun, Kagawa 761-0793, Japan bRare Sugar Research Center and Faculty of Agriculture, Kagawa University, Miki-cho, Kagawa 761-0795, Japan article info abstract Article history: L-Rhamnose isomerase (L-RhI) catalyzes the reversible isomerization of L-rhamnose to L-rhamnulose. Received 27 November 2012 Previously determined X-ray structures of L-RhI showed a hydride-shift mechanism for the isomeriza- Accepted 30 November 2012 tion of substrates in a linear form, but the mechanism for opening of the sugar-ring is still unclear. To elucidate this mechanism, we determined X-ray structures of a mutant L-RhI in complex with L- Keywords: rhamnopyranose and D-allopyranose. Results suggest that a catalytic water molecule, which acts as an l-Rhamnose isomerase acid/base catalyst in the isomerization reaction, is likely to be involved in pyranose-ring opening, and Pseudomonas stutzeri Sugar-ring opening mechanism that a newly found substrate sub-binding site in the vicinity of the catalytic site may recognize different Rare sugar anomers of substrates. X-ray structure C 2012 The Authors. -

Genomic Analysis of Thermophilicbacillus Coagulans
OPEN Genomic analysis of thermophilic Bacillus SUBJECT AREAS: coagulans strains: efficient producers for COMPARATIVE GENOMICS platform bio-chemicals BACTERIAL GENOMICS Fei Su & Ping Xu Received 17 September 2013 State Key Laboratory of Microbial Metabolism, and School of Life Sciences & Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, P. R. China. Accepted 14 January 2014 Microbial strains with high substrate efficiency and excellent environmental tolerance are urgently needed Published for the production of platform bio-chemicals. Bacillus coagulans has these merits; however, little genetic 29 January 2014 information is available about this species. Here, we determined the genome sequences of five B. coagulans strains, and used a comparative genomic approach to reconstruct the central carbon metabolism of this species to explain their fermentation features. A novel xylose isomerase in the xylose utilization pathway was identified in these strains. Based on a genome-wide positive selection scan, the selection pressure on amino Correspondence and acid metabolism may have played a significant role in the thermal adaptation. We also researched the requests for materials immune systems of B. coagulans strains, which provide them with acquired resistance to phages and mobile should be addressed to genetic elements. Our genomic analysis provides comprehensive insights into the genetic characteristics of P.X. ([email protected]. B. coagulans and paves the way for improving and extending the uses of this species. cn) hite biotechnology, the clean industrial technology supported by several predominant political move- ments, will comprise no less than 20% of the chemical industry sales in the United States in 20201.To attempt to meet the dramatic future demands for these materials, researchers have used microbial W 2 strains to produce platform bio-chemicals . -
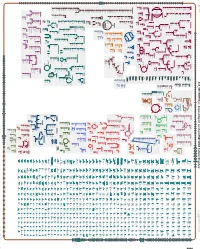
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114035Cyc: Amycolatopsis sacchari DSM 44468 Cellular Overview Connections between pathways are omitted for legibility. -

WO 2014/207087 Al 31 December 2014 (31.12.2014) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2014/207087 Al 31 December 2014 (31.12.2014) P O P C T (51) International Patent Classification: Seville (ES). GUTIERREZ GOMEZ, Pablo; Campus C12N 9/00 (2006.01) C12N 9/90 (2006.01) Palmas Altas, Calle Energia Solar 1, E-41014 Seville (ES). C12N 9/02 (2006.01) C12P 5/02 (2006.01) (74) Agent: NEDERLANDSCH OCTROOIBUREAU; J.W. C12N 9/10 (2006.01) C12P 7/76 (2006.01) Frisolaan 13, NL-2517 JS The Hague (NL). C12N 9/12 (2006.01) C12P 7/64 (2006.01) C12N 9/88 (2006.01) C12N 1/1 9 (2006.01) (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, (21) International Application Number: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, PCT/EP2014/063484 BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, (22) International Filing Date: DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, 26 June 2014 (26.06.2014) HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, (25) Filing Language: English MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, (26) Publication Language: English OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, (30) Priority Data: TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, 13382241 .1 26 June 2013 (26.06.2013) EP ZW. -

Characterization of Ribose-5-Phosphate Isomerase B from Newly Isolated Strain Ochrobactrum Sp
J. Microbiol. Biotechnol. (2018), 28(7), 1122–1132 https://doi.org/10.4014/jmb.1802.02021 Research Article Review jmb Characterization of Ribose-5-Phosphate Isomerase B from Newly Isolated Strain Ochrobactrum sp. CSL1 Producing L-Rhamnulose from L-Rhamnose Min Shen1†, Xin Ju1†, Xinqi Xu2, Xuemei Yao1, Liangzhi Li1*, Jiajia Chen1, Cuiying Hu1, Jiaolong Fu1, and Lishi Yan1 1School of Chemistry, Biology, and Material Engineering, Suzhou University of Science and Technology, Suzhou 215009, P.R. China 2Fujian Key Laboratory of Marine Enzyme Engineering, Fuzhou University, Fujian 350116, P.R. China Received: February 14, 2018 Revised: April 12, 2018 In this study, we attempted to find new and efficient microbial enzymes for producing rare Accepted: April 13, 2018 sugars. A ribose-5-phosphate isomerase B (OsRpiB) was cloned, overexpressed, and First published online preliminarily purified successfully from a newly screened Ochrobactrum sp. CSL1, which could May 8, 2018 catalyze the isomerization reaction of rare sugars. A study of its substrate specificity showed *Corresponding author that the cloned isomerase (OsRpiB) could effectively catalyze the conversion of L-rhamnose to Phone: +86-512-68056493; L-rhamnulose, which was unconventional for RpiB. The optimal reaction conditions (50oC, Fax: +86-512-68418431; 2+ E-mail: [email protected] pH 8.0, and 1 mM Ca ) were obtained to maximize the potential of OsRpiB in preparing L-rhamnulose. The catalytic properties of OsRpiB, including Km, kcat, and catalytic efficiency † These authors contributed (k /K ), were determined as 43.47 mM, 129.4 sec-1, and 2.98 mM/sec. The highest conversion equally to this work. -

Heat Priming Induces Trans-Generational Tolerance to High Temperature Stress in Wheat
fpls-07-00501 April 12, 2016 Time: 15:59 # 1 View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Frontiers - Publisher Connector ORIGINAL RESEARCH published: 14 April 2016 doi: 10.3389/fpls.2016.00501 Heat Priming Induces Trans-generational Tolerance to High Temperature Stress in Wheat Xiao Wang1, Caiyun Xin1,2, Jian Cai1, Qin Zhou1*, Tingbo Dai1, Weixing Cao1 and Dong Jiang1* 1 National Technology Innovation Center for Regional Wheat Production/National Engineering and Technology Center for Information Agriculture/Key Laboratory of Crop Physiology and Ecology in Southern China, Ministry of Agriculture, Nanjing Agricultural University, Nanjing, China, 2 Rice Research Institute, Shandong Academy of Agricultural Sciences, Jinan, China Wheat plants are very sensitive to high temperature stress during grain filling. Effects of heat priming applied to the first generation on tolerance of the successive generation to post-anthesis high temperature stress were investigated. Compared with the progeny of non-heat primed plants (NH), the progeny of heat-primed plants (PH) possessed higher grain yield, leaf photosynthesis and activities of antioxidant enzymes and lower cell membrane damage under high temperature stress. In the transcriptome profile, 1430 probes showed obvious difference in expression between PH and NH. These Edited by: genes were related to signal transduction, transcription, energy, defense, and protein Shabir Hussain Wani, destination and storage, respectively. The gene encoding the lysine-specific histone Sher-e-Kashmir University demethylase 1 (LSD1) which was involved in histone demethylation related to epigenetic of Agricultural Sciences and Technology, India modification was up-regulated in the PH compared with NH. -

B Number Gene Name Strand Orientation Protein Length Mrna
list list sample) short list predicted B number Operon ID Gene name assignment Protein length mRNA present mRNA intensity Gene description Protein detected - Strand orientation Membrane protein detected (total list) detected (long list) membrane sample Proteins detected - detected (short list) # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides Functional category detected (membrane Protein detected - total Protein detected - long b0001 thrL + 21 1344 P 1 0 0 0 0 thr operon leader peptide Metabolism of small molecules 1 b0002 thrA + 820 13624 P 39 P 18 P 18 P 18 P(m) 2 aspartokinase I, homoserine dehydrogenase I Metabolism of small molecules 1 b0003 thrB + 310 6781 P 9 P 3 3 P 3 0 homoserine kinase Metabolism of small molecules 1 b0004 thrC + 428 15039 P 18 P 10 P 11 P 10 0 threonine synthase Metabolism of small molecules 1 b0005 b0005 + 98 432 A 5 0 0 0 0 orf, hypothetical protein Open reading frames 2 b0006 yaaA - 258 1047 P 11 P 1 2 P 1 0 orf, hypothetical protein Open reading frames 3 b0007 yaaJ - 476 342 P 8 0 0 0 0 MP-GenProt-PHD inner membrane transport protein Miscellaneous 4 b0008 talB + 317 20561 P 20 P 13 P 16 P 13 0 transaldolase B Metabolism of small molecules 5 b0009 mog + 195 1296 P 7 0 0 0 0 required for the efficient incorporation of molybdate into molybdoproteins Metabolism of small molecules 6 b0010 yaaH - 188 407 A 2 0 0 0 0 PHD orf, hypothetical protein Open reading frames 7 b0011 b0011 - 237 338 P 13 0 0 0 0 putative oxidoreductase Miscellaneous 8 b0012 htgA -

Optimization of Fermentation Process of Pomegranate Peel And
molecules Article Optimization of Fermentation Process of Pomegranate Peel and Schisandra Chinensis and the Biological Activities of Fermentation Broth: Antioxidant Activity and Protective Effect Against H2O2-induced Oxidative Damage in HaCaT Cells Hui-Min Liu 1,2, Peng-Fei Xu 1, Ming-Yan Cheng 1, Sheng-Nan Lei 1, Qing-Lei Liu 1 and Wei Wang 1,2,* 1 School of Perfume & Aroma and Cosmetics, Shanghai Institute of Technology, Shanghai 201418, China; [email protected] (H.-M.L.); [email protected] (P.-F.X.); [email protected] (M.-Y.C.); [email protected] (S.-N.L.); [email protected] (Q.-L.L.) 2 Engineering Research Center of Perfume & Aroma and Cosmetics, Ministry of Education, Shanghai 201418, China * Correspondence: [email protected]; Tel.: +86-18918830550 Abstract: In this study, the lactobacillus fermentation process of pomegranate (Punica granatum L.) peel and Schisandra chinensis (Turcz.) Baill (PP&SC) was optimized by using the response surface method (RSM) coupled with a Box-Behnken design. The optimum fermentation condition with the maximal yield of ellagic acid (99.49 ± 0.47 mg/g) was as follows: 1:1 (w:w) ratio of pomegranate Citation: Liu, H.-M.; Xu, P.-F.; peel to Schisandra chinensis, 1% (v:v) of strains with a 1:1 (v:v) ratio of Lactobacillus Plantarum to Cheng, M.-Y.; Lei, S.-N.; Liu, Q.-L.; Streptococcus Thermophilus, a 37 ◦C fermentation temperature, 33 h of fermentation time, 1:20 (g:mL) Wang, W. Optimization of of a solid–liquid ratio and 3 g/100 mL of a glucose dosage. -

Production, Optimization and Characterization of Α-Amylase and Glucose Isomerase Producing Bacillus Megaterium BPTK5 from Cassava Waste
Available online a t www.pelagiaresearchlibrary.com Pelagia Research Library European Journal of Experimental Biology, 2012, 2 (3):590-595 ISSN: 2248 –9215 CODEN (USA): EJEBAU Production, optimization and characterization of α-amylase and glucose isomerase producing Bacillus megaterium BPTK5 from cassava waste D. J. Mukesh kumar a*, T. Silambarasan b, R. Renugac, M. Ravi kumar a, S. Karthigai devi c, Dhandapani Ramamurthy b and P. T. Kalaichelvan a aCentre for Advanced Studies in Botany, University of Madras, Guindy campus, Chennai, TN, India bDepartment of Microbiology, Periyar University, Salem, TN, India cK.M.G. College of Arts and Science, Gudiyatham, TN, India _____________________________________________________________________________ ABSTRACT A mesophilic bacterium, Bacillus megaterium BPTK5 was isolated from cassava waste samples collected from starch processing plant (TN, India). The isolate was assayed for its ability to produce α-amylase and glucose isomerase. Experiments were set to observe the effects of incubation time, pH, temperature, and carbon and nitrogen sources on the growth of BPTK5 and its enzyme activities. Optimum biomass and amylolytic activity was achieved after 76 h of incubation, at 35°C, pH 6.0. Among the various carbon and nitrogen sources investigated, starch and casein were found to be the best inducers of α-amylase. An increase in glucose isomerase activity was observed at 48 h of incubation at 35°C with the initial pH of 6in the presence of Xylose and peptone. Thus Bacillus megaterium BPTK5 was proved to be an industrially important microbe involved in HFCS manufacturing. Key Words: Starch, Casein, CMC column, HFCS, Bacillus megaterium . _____________________________________________________________________________ INTRODUCTION In most of the food industries, Carbohydrate sweeteners were used for their sweetness when compared to conventional sweeteners.