Urea-Cycle Disorders As a Paradigm for Inborn Errors of Hepatocyte Metabolism
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Leading Article the Molecular and Genetic Base of Congenital Transport
Gut 2000;46:585–587 585 Gut: first published as 10.1136/gut.46.5.585 on 1 May 2000. Downloaded from Leading article The molecular and genetic base of congenital transport defects In the past 10 years, several monogenetic abnormalities Given the size of SGLT1 mRNA (2.3 kb), the gene is large, have been identified in families with congenital intestinal with 15 exons, and the introns range between 3 and 2.2 kb. transport defects. Wright and colleagues12 described the A single base change was identified in the entire coding first, which concerns congenital glucose and galactose region of one child, a finding that was confirmed in the malabsorption. Subsequently, altered genes were identified other aZicted sister. This was a homozygous guanine to in partial or total loss of nutrient absorption, including adenine base change at position 92. The patient’s parents cystinuria, lysinuric protein intolerance, Menkes’ disease were heterozygotes for this mutation. In addition, it was (copper malabsorption), bile salt malabsorption, certain found that the 92 mutation was associated with inhibition forms of lipid malabsorption, and congenital chloride diar- of sugar transport by the protein. Since the first familial rhoea. Altered genes may also result in decreased secretion study, genomic DNA has been screened in 31 symptomatic (for chloride in cystic fibrosis) or increased absorption (for GGM patients in 27 kindred from diVerent parts of the sodium in Liddle’s syndrome or copper in Wilson’s world. In all 33 cases the mutation produced truncated or disease)—for general review see Scriver and colleagues,3 mutant proteins. -

What I Tell My Patients About Cystinuria
BRITISH JOURNAL OF RENAL MEDICINE 2016; Vol 21 No 1 Patient information John A Sayer MB ChB FRCP PhD Consultant What I tell my patients Nephrologist1,2 Charles Tomson MA BM BCh FRCP DM Consultant Nephrologist2 about cystinuria 1 Institute of Genetic Medicine, Newcastle Cystinuria is an inherited condition that causes kidney stones in children and adults. University 2 Renal Services, Newcastle upon Tyne John A Sayer and Charles Tomson describe the effects of this condition and how to Hospitals NHS Foundation Trust, manage it successfully. Freeman Hospital, Newcastle upon Tyne Cystine is an amino acid found in high- Box 1. You say stones… I say calculi protein foods such as meat, eggs and dairy. High concentrations of cystine, particularly ‘Kidney stone’ and ‘renal calculus’ means the same thing – a solid piece of material that forms in the in acidic urine, result in crystallisation of kidneys. The word ‘calculus’ is derived from Latin, cystine, leading to the formation of kidney literally meaning ‘small pebble’, as used on an stones (see Box 1). These cystine stones are a abacus; the plural of calculus is calculi. ‘Nephrolith’ rare form of kidney stone, accounting for is another name for a kidney stone around 6% of kidney stones in children and around 1% of those in adults.1 Cystinuria is inherited in different ways and this can Cystinuria is estimated to affect 1 in 7,000 people.2 be confusing. Most patients have to inherit two faulty Despite the condition being present from birth, most copies of the gene involved (one inherited from their people affected will get their first stone in their mother and one from their father) to be affected by twenties, although a quarter of patients present in the condition. -

Amyloid Like Aggregates Formed by the Self-Assembly of Proline And
Please do not adjust margins Journal Name ARTICLE Amyloid like aggregates formed by the self-assembly of proline and Hydroxyproline Bharti Koshtia, Ramesh Singh Chilwalb, Vivekshinh Kshtriyaa, Shanka Walia c, Dhiraj Bhatiac, K.B. Joshib* and Nidhi Goura* a Department of Chemistry, Indrashil University, Mehsana, Gujarat, India b Department of Chemistry, Dr. Hari Singh Gour, Sagar University, Madhya Pradesh, India c Biological Engineering Discipline, Indian Institute of Technology Gandhinagar, Gujarat, India Abstract: Single amino acid based self-assembled structures have gained a lot of interest recently owing to their pathological significance in metabolite disorders. There is plethora of significant research work which illustrate amyloid like characteristics of assemblies formed by aggregation of single amino acids like Phenylalanine, Tyrosine, Tryptophan, Cysteine and Methionine and its implications in pathophysiology of single amino acid metabolic disorders like phenylketonuria, tyrosinemia, hypertryptophanemia, cystinuria and hypermethioninemia respectively. Hence, studying aggregation behaviour of single amino acids is very crucial to assess the underlying molecular mechanism behind metabolic disorders. In this manuscript we report for the very first time the aggregation properties of non-aromatic single amino acids Hydroxy-proline and Proline. The morphologies of these were studied extensively by Optical microscopy (OM), ThT binding fluorescence microscopy, Scanning Electron Microscopy (SEM) and Atomic force microscopy (AFM). It can be assessed that these amino acids form globular structures at lower concentrations and gradually changes to tape like structures on increasing the This journal is © The Royal Society of Chemistry 20xx J. Name., 2013, 00, 1-3 | 1 Please do not adjust margins Please do not adjust margins Journal Name ARTICLE concentration as assessed by AFM. -
ACVIM Giger Cyst+Fanconi 2014F
UPDATES ON CYSTINURIA AND FANCONI SYNDROME: AMINO ACIDURIAS IN DOGS Urs Giger, DACVIM-SA, DECVIM CA, DECVCP, Ann-Kathrin Brons, Caitlin A Fitzgerald, Jeffrey Slutsky, Karthik Raj, Victor Stora, Adrian C Sewell and Paula S Henthorn Philadelphia, PA Introduction Disorders of the renal proximal tubules can cause selective or generalized aminoaciduria and may be associated with urinary losses of other solutes such as glucose, lactate, electrolytes and bicarbonate. Two renal tubular defects involving amino acids have long been recognized in dogs, namely cystinuria, leading to cystine calculi and urinary obstruction, and Fanconi syndrome, progressing to renal failure if untreated. Both hereditary disorders have been investigated at the molecular level and are more complex than originally anticipated. Furthermore, the ingestion of Chinese jerky treats has recently been found to be associated with Fanconi syndrome in many dogs and rarely cats. The current understanding of pathophysiology, clinicopathological findings, diagnosis, and therapeutic options will be presented. Fanconi Syndrome Fanconi syndrome, named after the Swiss pediatrician Guido Fanconi and also known as Fanconi’s syndrome or Fanconi disease, should not be confused with Fanconi anemia, a bone marrow disorder in humans. Fanconi syndrome represents a majorproximal renal tubular defect, which hampers the adequate reabsorption of glucose, amino acids, bicarbonate, sodium, calcium, phosphate, lactate, ketones, and carnitine. This rather general loss of multiple functions of the proximal renal tubules can be associated with renal tubular acidosis and lead to progressive renal failure if left untreated. In the renal tubules there are multiple co- transporters for sodium and glucose, amino acids, calcium, and inorganic phosphorus and a sodium/hydrogen ion antiporter, which, depending upon the concentration gradient established by the sodium-potassium pump, move hydrogen ions into the urine. -

Cystinosis: Antibodies and Healthy Bodies
J Am Soc Nephrol 13: 2189–2191, 2002 Cystinosis: Antibodies and Healthy Bodies ROBERT KLETA AND WILLIAM A. GAHL Section on Human Biochemical Genetics, Heritable Disorders Branch, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland. Nephropathic cystinosis was first described in the early 1900s transplant cystinosis patients. At the same time, eyedrops con- in a 21-mo-old boy who died of progressive anorexia; two taining cysteamine (0.5%) were shown to dissolve the corneal siblings had previously died in infancy under similar circum- crystals, which cause a painful photophobia and occasional stances (1). By meticulous observations and analyses, it be- epithelial erosions (21-23). The crystals are pathognomic for came clear that abnormal cystine accumulation was character- cystinosis and can be identified by an experienced ophthalmol- istic of this autosomal recessive disease (2-4). Although some ogist as early as 1 yr of age (24). considered it to be a severe form of cystinuria, cystinosis was The era of molecular biology has brought with it an under- clearly distinguished from cystinuria by Bickel’s excellent standing of the genetic basis of cystinosis. In the mid 1990s, clinical and biochemical observations (5). Clinically, untreated the cystinosis gene was mapped to chromosome 17p (25); in cystinosis patients would suffer renal tubular Fanconi syn- 1998, the gene CTNS, coding for a lysosomal transport protein drome, with hypophosphatemic rickets, hypokalemia, polyuria, named cystinosin, was isolated (26). A 57,257-bp deletion (27) polydypsia, dehydration, acidosis, and growth retardation fol- was found to be responsible for approximately half of Northern lowed by end-stage renal disease (ESRD) and death at approx- European and North American cystinosis patients (26,28); this imately 10 yr of age (6,7). -

The PAH Gene, Phenylketonuria, and a Paradigm Shift
HUMAN MUTATION 28(9), 831^845, 2007 WILEY 200TH ANNIVERSARY TRIBUTE ARTICLE The PAH Gene, Phenylketonuria, and a Paradigm Shift Charles R. Scriver1–4Ã 1Department of Human Genetics, Faculty of Medicine, McGill University, Montreal, Quebec, Canada; 2Department of Biochemistry, Faculty of Medicine, McGill University, Montreal, Quebec, Canada; 3Department of Pediatrics, Faculty of Medicine, McGill University, Montreal, Quebec, Canada; 4Department of Biology, Faculty of Science, McGill University, Montreal, Quebec, Canada Communicated by Johannes Zschocke ‘‘Inborn errors of metabolism,’’ first recognized 100 years ago by Garrod, were seen as transforming evidence for chemical and biological individuality. Phenylketonuria (PKU), a Mendelian autosomal recessive phenotype, was identified in 1934 by Asbjo¨rn Fo¨lling. It is a disease with impaired postnatal cognitive development resulting from a neurotoxic effect of hyperphenylalaninemia (HPA). Its metabolic phenotype is accountable to multifactorial origins both in nurture, where the normal nutritional experience introduces L-phenylalanine, and in nature, where mutations (4500 alleles) occur in the phenylalanine hydroxylase gene (PAH) on chromosome 12q23.2 encoding the L-phenylalanine hydroxylase enzyme (EC 1.14.16.1). The PAH enzyme converts phenylalanine to tyrosine in the presence of molecular oxygen and catalytic amounts of tetrahydrobiopterin (BH4), its nonprotein cofactor. PKU is among the first of the human genetic diseases to enter, through newborn screening, the domain of public health, and to show a treatment effect. This effect caused a paradigm shift in attitudes about genetic disease. The PKU story contains many messages, including: a framework on which to appreciate the complexity of PKU in which phenotype reflects both locus-specific and genomic components; what the human PAH gene tells us about human population genetics and evolution of modern humans; and how our interest in PKU is served by a locus-specific mutation database (http://www.pahdb.mcgill.ca; last accessed 20 March 2007). -

Alkaptonuria
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Repositório Aberto da Universidade do Porto Alkaptonuria An obscure disease JOANA PEREIRA DA SILVA LAMAS DISSERTAÇÃO DE MESTRADO INTEGRADO EM MEDICINA 2016 [ALKAPTONURIA] P a g e | 2 Autor: Joana Pereira da Silva Lamas Orientador: Sara Isabel Mendes Rocha Assistente hospitalar de Medicina Interna no Centro Hospitalar do Porto – Hospital de Santo António Afiliação: Instituto de Ciências Biomédicas Abel Salazar Rua de Jorge Viterbo Ferreira nº 228 4050-313 PORTO [ALKAPTONURIA] P a g e | 3 ÍNDICE Abstract ..............................................................................................................................5 Resumo..............................................................................................................................6 Introduction ........................................................................................................................7 Materials and Methods ......................................................................................................8 History ................................................................................................................................9 Genetics and Metabolic Pathway ....................................................................................10 Epidemiology ...................................................................................................................13 Clinical Features and Natural History .............................................................................14 -

Early Diagnosis of Classic Homocystinuria in Kuwait Through Newborn Screening: a 6-Year Experience
International Journal of Neonatal Screening Article Early Diagnosis of Classic Homocystinuria in Kuwait through Newborn Screening: A 6-Year Experience Hind Alsharhan 1,2,3,*, Amir A. Ahmed 4,5 , Naser M. Ali 5 , Ahmad Alahmad 6, Buthaina Albash 3, Reem M. Elshafie 3,5, Sumaya Alkanderi 3,5, Usama M. Elkazzaz 7, Parakkal Xavier Cyril 8, Rehab M. Abdelrahman 4, Alaa A. Elmonairy 3, Samia M. Ibrahim 9, Yasser M. E. Elfeky 10, Doaa I. Sadik 3, Sara D. Al-Enezi 6, Ayman M. Salloum 11, Yadav Girish 12, Mohammad Al-Ali 5, Dina G. Ramadan 13, Rasha Alsafi 14, May Al-Rushood 4 and Laila Bastaki 3 1 Department of Pediatrics, Faculty of Medicine, Kuwait University, P.O. Box 24923, Safat 13110, Kuwait 2 Department of Pediatrics, Farwaniya Hospital, Ministry of Health, Sabah Al-Nasser 92426, Kuwait 3 Kuwait Medical Genetics Center, Ministry of Health, Sulaibikhat 80901, Kuwait; [email protected] (B.A.); [email protected] (R.M.E.); [email protected] (S.A.); [email protected] (A.A.E.); [email protected] (D.I.S.); [email protected] (L.B.) 4 Newborn Screening Laboratory, Kuwait Medical Genetics Center, Ministry of Health, Sulaibikhat 80901, Kuwait; [email protected] (A.A.A.); [email protected] (R.M.A.); [email protected] (M.A.-R.) 5 Next Generation Sequencing Laboratory, Kuwait Medical Genetics Center, Ministry of Health, Sulaibikhat 80901, Kuwait; [email protected] (N.M.A.); [email protected] (M.A.-A.) 6 Molecular Genetics Laboratory, Kuwait Medical Genetics Center, Ministry of Health, Sulaibikhat 80901, Kuwait; -
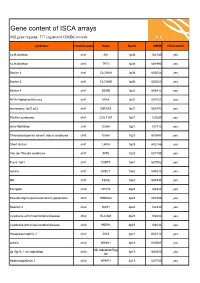
ISCA Disease List
Gene content of ISCA arrays 402 gene regions, 377 registered OMIM records syndrome chromosome Gene Band OMIM ISCA 8x60k 1p36 deletion chr1 SKI 1p36 164780 yes 1p36 deletion chr1 TP73 1p36 601990 yes Bartter 4 chr1 CLCNKA 1p36 602024 yes Bartter 3 chr1 CLCNKB 1p36 602023 yes Bartter 4 chr1 BSND 1p32 606412 yes NFIA Haploinsufficiency chr1 NFIA 1p31 600727 yes monosomy 1p31 p22 chr1 DIRAS3 1p31 605193 yes Stickler syndrome chr1 COL11A1 1p21 120280 yes atrial fibrillation chr1 GJA5 1q21 121013 yes Thrombocytopenia absent radius syndrome chr1 GJA8 1q21 600897 yes Short stature chr1 LHX4 1q25 602146 yes Van der Woude syndrome chr1 IRF6 1q32 607199 yes Fryns 1q41 chr1 DISP1 1q41 607502 yes autism chr1 DISC1 1q42 605210 yes MR chr1 TBCE 1q42 604934 yes Feingold chr2 MYCN 2q24 164840 yes Pseudovaginal perineoscrotal hypospadias chr2 SRD5A2 2p23 607306 yes Noonan 4 chr2 SOS1 2p22 182530 yes Cystinuria with mitochondrial disease chr2 SLC3A1 2p21 104614 yes Cystinuria with mitochondrial disease chr2 PREPL 2p21 104614 yes Holopresencaphly 2 chr2 SIX3 2p21 603714 yes autism chr2 NRXN1 2p16 600565 yes MicrodeletionReg 2p15p16.1 microdeletion chr2 2p15 602559 yes ion Nephronophthsis 1 chr2 NPHP1 2q13 607100 yes Holoprosencephaly 9 chr2 GLI2 2q14 165230 yes visceral heterotaxy chr2 CFC1 2q21 605194 yes Mowat-Wilson syndrome chr2 ZEB2 2q22 605802 yes autism chr2 SLC4A10 2q24 605556 yes SCN1A-related seizures chr2 SCN1A 2q24 182389 yes HYPOMYELINATION, GLOBAL CEREBRAL chr2 SLC25A12 2q31 603667 yes Split/hand foot malformation -5 chr2 DLX1 2q31 600029 yes -

Leukocyte and Liver Glutaminase in Lysinuric Protein Intolerance
Pediat. Res. 6: 797-801 (1972) Amino acid metabolism leukocyte ammonia liver glutaminase urea cycle Leukocyte and Liver Glutaminase in Lysinuric Protein Intolerance O. SlMELL(26], J. PERHEENTUPA, AND J. K. VlSAKORPI University of Helsinki Children's Hospital, Helsinki, Finland Extract Leukocyte glutaminase / activity was studied in 17 patients with lysinuric protein intolerance (LPI, familial protein intolerance) and 21 controls. The values were logarithmically distributed, the mean (95% confidence interval) being 9.1 (1.5-54.3) for the patients and 15.4 (2.3-104.4) for the controls, in nanomoles of ammonia per 106 leukocytes and 30 min. The difference is significant at P < 0.05. Only two of the LPI patients had a value below the range of the controls, and five others actually had a value above the mean of the controls. Liver glutaminase / activity was measured in two of the LPI patients and in seven controls. The activity found in the patients was clearly higher than in the controls. We have disproved the speculation of a deficiency of liver glutaminase in LPI, as a basic derangement in LPI, common to botr basic chemical disturbances of LPI, the renal leakage of basic amino acids and the delayed urea synthesis. Speculation Patients with LPI have decreased plasma levels of arginine, ornithine, and lysine. Arginine and ornithine supplement accelerates their slow urea synthesis. The possi- bility that this deficiency of arginine and ornithine is further exaggerated through a defect of their transport to the site of urea production in the parenchymal cells of liver is currently under investigation. -
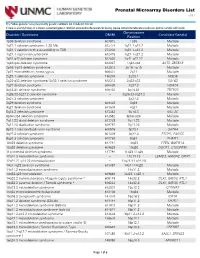
Prenatal Microarray Disorders List V19.1
Prenatal Microarray Disorders List v19.1 This "whole genome" array may identify genetic conditions not included in this list. If there is a family history of a known suspected genetic condition unrelated to the reason for testing, please contact the laboratory to discuss prior to sample submission. Chromosome Disorder / Syndrome OMIM Candidate Gene(s) Position 1p36 deletion syndrome 607872 1p36 Multiple 1q21.1 deletion syndrome, 1.35 Mb 612474 1q21.1-q21.2 Multiple 1q21.1 deletion with susceptibility to TAR 274000 1q21.1-q21.2 Multiple 1q21.1 duplication syndrome 612475 1q21.1-q21.2 Multiple 1q41-q42 deletion syndrome 612530 1q41-q42.12 Multiple 1q43-q44 deletion syndrome 612337 1q43-q44 AKT3, ZBTB18 2p16.1-p15 deletion syndrome 612513 2p16.1-p15 Multiple 2p21 microdeletion, homozygous 606407 2p21 Multiple 2q23.1 deletion syndrome 156200 2q23.1 MBD5 2q32-q33 deletion syndrome/ 2q33.1 deletion syndrome 612313 2q32-q33 SATB2 2q37 deletion syndrome 600430 2q37.3 HDAC4 3q13.31 deletion syndrome 615433 3q13.31 ZBTB20 3q26.33-3q27.2 deletion syndrome -- 3q26.33-3q27.2 Multiple 3q27.3 deletion syndrome -- 3q27.3 Multiple 3q29 deletion syndrome 609425 3q29 Multiple 4q21 deletion syndrome 613509 4q21 Multiple 5q14.3 deletion syndrome 613443 5q14.3 MEF2C 6pter-p24 deletion syndrome 612582 6pter-p24 Multiple 7q11.23 distal deletion syndrome 613729 7q11.23 Multiple 7q11.23 duplication syndrome 609757 7q11.23 Multiple 8p23.1 deletion/duplication syndrome 600576 8p23.1 GATA4 9q22.3 deletion syndrome 601309 9q22.3 PTCH1, FANCC 9q34.3 deletion syndrome -

Supplemental Canine Cystine Information
Minnesota Urolith Center UNIVERSITY OF MINNESOTA College of Veterinary Medicine 1352 Boyd Avenue St Paul, MN 55108 Urolithcenter.org Phone 612.625.4221 Fax 612.626.3226 email [email protected] Common Questions on Dogs Forming Cystine Stones 10/2019 How will I know if my dog has cystinuria? 1. Cystine is an amino acid that is freely filtered in urine and almost completely reabsorbed by the kidney tubules (i.e. removed from the urine). 2. As urine cystine concentration increases, a positive urine nitroprusside test, cystine crystals and cystine stones are indicators of disease. 3. Although cystinuria and cystine crystalluria do not cause clinical signs, cystine stones can irritate the lining of the urinary tract resulting in urinary accidents, urgency, straining, or bloody urine. In some cases, the stones result in life-threatening urinary obstruction. Cystine stones are not always visible on x-rays and may require special contrast studies or an ultrasound to diagnosis. 4. Genetic tests for cystinuria are available for some breeds of dogs. A genetic marker test for androgen dependent cystinuria has been developed for Mastiffs, English bulldogs and French bulldogs. There are other breeds with androgen dependent cystinuria as well as other breeds in which castration will not reduce cystinuria (Type Ia, reported in Newfoundland dogs, Labradors and Landseers,Type IIa, reported in Australian Cattle dogs and Border Collies, Type IIb, reported in Miniature Pinschers). Will castration prevent recurrence of cystine stones? 1. Surgical or medical castration can resolve/cure cystinuria in the subset of male dogs with androgen dependent cystinuria. 2. The magnitude of cystinuria associated with stone formation is wide (100 to >10,000 ɥmol/g creatinine) and varies between serial measurements in the same dog.