Non-Cell-Based in Vitro Methods in the Study of Prodrug Absorption and Metabolism
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reduction of Pectinesterase Activity in a Commercial Enzyme Preparation
Journal of the Science of Food and Agriculture J Sci Food Agric 85:1613–1621 (2005) DOI: 10.1002/jsfa.2154 Reduction of pectinesterase activity in a commercial enzyme preparation by pulsed electric fields: comparison of inactivation kinetic models Joaquın´ Giner, Pascal Grouberman, Vicente Gimeno and Olga Martın´ ∗ Department of Food Technology, University of Lleida, CeRTA-UTPV, ETSEA, Avda Alcalde Rovira Roure 191, 25198-Lleida, Spain Abstract: The inactivation of pectinesterase (PE) in a commercial enzyme preparation (CEP) under high intensity pulsed electric fields (HIPEF) was studied. After desalting and water dilution of the raw CEP, samples were exposed to exponentially decay waveform pulses for up to 463 µs at electric field intensities ranging from 19 to 38 kV cm−1. Pulses were applied in monopolar mode. Experimental data were fitted to a first-order kinetic model as well as to models based on Fermi, Hulsheger¨ or Weibull equations to describe PE inactivation kinetics. Characteristic parameters for each model were calculated. Relationships between some of the parameters and process variables were obtained. The Weibull model yielded the best accuracy factor. The relationship between residual PE and input of electrical energy density was found to be that of exponential decay. 2005 Society of Chemical Industry Keywords: pulsed electric fields; kinetics; pectinesterase; model; inactivation INTRODUCTION It has become customary to use CEPs in fruit and Pectinesterase (PE; EC 3.1.1.11) is a pectic enzyme vegetable juice technology. Depending -

Download Product Insert (PDF)
PRODUCT INFORMATION Lysophospholipase D Polyclonal Antibody Item No. 10005375 Overview and Properties Contents: This vial contains 500 µl of peptide affinity-purified antibody. Synonyms: Autotaxin, ENPP2, lysoPLD Immunogen: Peptide from the C-terminal region of rat LysoPLD Species Reactivity: (+) Human, mouse, and rat; other species not tested Form: Liquid Storage: -20°C (as supplied) Stability: ≥1 year Storage Buffer: TBS, pH 7.4, with 50% glycerol, 0.1%l BSA, and 0.02% sodium azide Host: Rabbit Applications: Immunocytochemistry (ICC), Immunohistochemistry (IHC), and Western blot (WB); the recommended starting dilution for ICC is 1:500, 1:80 for IHC, and 1:200 for WB. Other applications were not tested, therefore optimal working concentration/dilution should be determined empirically. Images 1 · · · · · · · 104 kDa · · · · · · · 60 kDa Lane 1: Human cerebella supernatant (40 µg) Immunohistochemistry analysis of formalin-fixed, paraffin-embedded (FFPE) human cerebellum ǎssue aer heat-induced anǎgen retrieval in pH 6.0 citrate buffer. Aer incubaǎon with Lysophospholipase D Polyclonal Anǎbody (Item No. 10005375) at a 1:80 diluǎon, slides were incubated with bioǎnylated secondary anǎbody, followed by alkaline phosphatase-strepavidin and chromogen (DAB). WARNING CAYMAN CHEMICAL THIS PRODUCT IS FOR RESEARCH ONLY - NOT FOR HUMAN OR VETERINARY DIAGNOSTIC OR THERAPEUTIC USE. 1180 EAST ELLSWORTH RD SAFETY DATA ANN ARBOR, MI 48108 · USA This material should be considered hazardous until further information becomes available. Do not ingest, inhale, get in eyes, on skin, or on clothing. Wash thoroughly after handling. Before use, the user must review the complete Safety Data Sheet, which has been sent via email to your institution. PHONE: [800] 364-9897 WARRANTY AND LIMITATION OF REMEDY [734] 971-3335 Buyer agrees to purchase the material subject to Cayman’s Terms and Conditions. -

Diagnostic Value of Serum Enzymes-A Review on Laboratory Investigations
Review Article ISSN 2250-0480 VOL 5/ ISSUE 4/OCT 2015 DIAGNOSTIC VALUE OF SERUM ENZYMES-A REVIEW ON LABORATORY INVESTIGATIONS. 1VIDYA SAGAR, M.SC., 2DR. VANDANA BERRY, MD AND DR.ROHIT J. CHAUDHARY, MD 1Vice Principal, Institute of Allied Health Sciences, Christian Medical College, Ludhiana 2Professor & Ex-Head of Microbiology Christian Medical College, Ludhiana 3Assistant Professor Department of Biochemistry Christian Medical College, Ludhiana ABSTRACT Enzymes are produced intracellularly, and released into the plasma and body fluids, where their activities can be measured by their abilities to accelerate the particular chemical reactions they catalyze. But different serum enzymes are raised when different tissues are damaged. So serum enzyme determination can be used both to detect cellular damage and to suggest its location in situ. Some of the biochemical markers such as alanine aminotransferase, aspartate aminotransferase, alkaline phasphatase, gamma glutamyl transferase, nucleotidase, ceruloplasmin, alpha fetoprotein, amylase, lipase, creatine phosphokinase and lactate dehydrogenase are mentioned to evaluate diseases of liver, pancreas, skeletal muscle, bone, etc. Such enzyme test may assist the physician in diagnosis and treatment. KEYWORDS: Liver Function tests, Serum Amylase, Lipase, CPK and LDH. INTRODUCTION mitochondrial AST is seen in extensive tissue necrosis during myocardial infarction and also in chronic Liver diseases like liver tissue degeneration DIAGNOSTIC SERUM ENZYME and necrosis². But lesser amounts are found in Enzymes are very helpful in the diagnosis of brain, pancreas and lung. Although GPT is plentiful cardiac, hepatic, pancreatic, muscular, skeltal and in the liver and occurs only in the small amount in malignant disorders. Serum for all enzyme tests the other tissues. -
What Is Alkaline Phosphatase? What Are the Symptoms of Low ALP?
What is the treatment for HPP? In 2015, asfotase alfa (Strensiq™) was approved for use in the US, the European Union, and Canada for pediatric-onset HPP, and in Japan for HPP with onset at any age. The medication is an injection given multiple times per week underneath the skin (subcutaneous). It is a recombinant (factory-made) form of ALP that has a bone-targeting component. When patients take asfotase alfa, ALP levels measured in the blood are quite high (often in the multiple thousands). Therefore, measuring ALP levels is typically not helpful once a patient is on therapy. What is Alkaline Phosphatase? What are the symptoms of low ALP? For more information, please contact Patients with hypophosphatasia (HPP) have Low ALP can lead to multiple symptoms of HPP, the Soft Bones Foundation. a low blood alkaline phosphatase (ALP) level. including poor bone mineralization and rickets as well (866) 827-9937 – Toll Free as early tooth loss (prior to age 5 years). It is also (973) 453-3093 – Direct Line thought that PPi may build up in muscle tissue 121 Hawkins Place, #267 ALP is an enzyme (a protein that breaks down and may be responsible for the pain and muscle Boonton, New Jersey 07005 chemicals). Most health providers are aware that weakness that some patients with HPP experience. www.SoftBones.org high ALP levels may indicate medical problems, such as liver disease or other bone diseases besides HPP, but many providers may not recognize that low ALP levels can indicate HPP. Low ALP is important What questions should patients ask Written by because the inability to break down chemicals can their doctors about ALP? Jill Simmons, MD lead to elevated levels of these chemicals, which Associate Professor of Pediatrics, Ian Burr Division can cause multiple problems. -

Effect of Alkaline Phosphatase on the Function of Purified Reverse Transcriptase in Reconstructed Reverse Transcription (116 Pp) Director: Kenneth F
University of Montana ScholarWorks at University of Montana Graduate Student Theses, Dissertations, & Professional Papers Graduate School 1984 Effect of alkaline phosphatase on the function of purified er verse transcriptase in reconstructed reverse transcription Michael J. Wolkowicz The University of Montana Follow this and additional works at: https://scholarworks.umt.edu/etd Let us know how access to this document benefits ou.y Recommended Citation Wolkowicz, Michael J., "Effect of alkaline phosphatase on the function of purified er verse transcriptase in reconstructed reverse transcription" (1984). Graduate Student Theses, Dissertations, & Professional Papers. 7480. https://scholarworks.umt.edu/etd/7480 This Thesis is brought to you for free and open access by the Graduate School at ScholarWorks at University of Montana. It has been accepted for inclusion in Graduate Student Theses, Dissertations, & Professional Papers by an authorized administrator of ScholarWorks at University of Montana. For more information, please contact [email protected]. COPYRIGHT ACT OF 1976 This is an unpublished manuscript in which copyright sub s i s t s . Any further reprinting of its contents must be approved BY THE a u t h o r . Mansfield Library University of Montana Date ; 1 G 8 5 EFFECT OF ALKALINE PHOSPHATASE ON THE FUNCTION OF PURIFIED REVERSE TRANSCRIPTASE IN RECONSTRUCTED REVERSE TRANSCRIPTION By Michael J. Wolkowicz B. A. American International College, 1976 B. S. University of Montana, 1980 Presented in partial fulfillment of the requirements for the degree of Masters of Science UNIVERSITY OF MONTANA 1984 Approved by: iP- airman. Board of Examiners D^n, Graduater'school Date UMI Number: EP38281 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. -

The Characterization of Oligonucleotides and Nucleic Acids Using Ribonuclease H and Mass Spectrometry
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1999 The hC aracterization of Oligonucleotides and Nucleic Acids Using Ribonuclease H and Mass Spectrometry. Lenore Marie Polo Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Polo, Lenore Marie, "The hC aracterization of Oligonucleotides and Nucleic Acids Using Ribonuclease H and Mass Spectrometry." (1999). LSU Historical Dissertations and Theses. 6923. https://digitalcommons.lsu.edu/gradschool_disstheses/6923 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter free, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand corner and continuing from left to right in equal sections with small overlaps. -

Regulation of Fructose-6-Phosphate 2-Kinase By
Proc. Natt Acad. Sci. USA Vol. 79, pp. 325-329, January 1982 Biochemistry Regulation of fructose-6-phosphate 2-kinase by phosphorylation and dephosphorylation: Possible mechanism for coordinated control of glycolysis and glycogenolysis (phosphofructokinase) EISUKE FURUYA*, MOTOKO YOKOYAMA, AND KOSAKU UYEDAt Pre-Clinical Science Unit of the Veterans Administration Medical Center, 4500 South Lancaster Road, Dallas, Texas 75216; and Biochemistry Department of the University ofTexas Health Science Center, 5323 Harry Hines Boulevard, Dallas, Texas 75235 Communicated by Jesse C. Rabinowitz, September 28, 1981 ABSTRACT The kinetic properties and the control mecha- Fructose 6-phosphate + ATP nism of fructose-6-phosphate 2-kinase (ATP: D-fructose-6-phos- -3 Fructose + ADP. [1] phate 2-phosphotransferase) were investigated. The molecular 2,6-bisphosphate weight of the enzyme is -100,000 as determined by gel filtration. The plot of initial velocity versus ATP concentration is hyperbolic We have shown that the administration of extremely low con- with a K. of 1.2 mM. However, the plot of enzyme activity as a centrations of glucagon (0.1 fM) or high concentrations of epi- function of fructose 6-phosphate is sigmoidal. The apparent K0.5 nephrine (10 ,uM) to hepatocytes results in inactivation offruc- for fructose 6-phosphate is 20 ,IM. Fructose-6-phosphate 2-kinase tose-6-phosphate 2-kinase and concomitant decrease in the is inactivated by -the catalytic subunit of cyclic AMP-dependent fructose 2,6-bisphosphate level (12). These results, as well as protein kinase, and the inactivation is closely correlated with phos- more recent data using Ca2+ and the Ca2+ ionophore A23187 phorylation. -

Simple, Sensitive Detection of Protein Phosphodiesterase Activity Measure Real-Time Signal Differences in the Picomole Range with the Phosphate Sensor Method
APPLICATION NOTE Phosphodiesterase Activity: A Phophate Sensor Application Simple, Sensitive Detection of Protein Phosphodiesterase Activity Measure real-time signal differences in the picomole range with the Phosphate Sensor method Described in this application is a method utilizing the Life Technologies Phosphate Sensor, a simple tool to interrogate the activity of phosphate-releasing enzymes. The assays described detect the increase of fluorescence intensity when free inorganic phosphate binds to a bacterially derived phosphate-binding protein modified with a fluorophore.To evaluate the Phosphate Sensor methods, we compared detection by coupling to an alkaline phosphatase against a commonly used coupled luciferase assay, examined responses for human phosphodiesterase A (PTE5A) in a titration, in real-time kinetic mode, and with a sildenafil citrate inhibitor. Phosphate Sensor is orders of magnitude more sensitive than the coupled luciferase method, faster and simpler to use than other competitor methods, and uniquely qualified for determining enzymatic rates. Second messenger systems that involve an increase or decrease of cyclic nucleotides (cAMP or cGMP) mediate intracellular signal trans- duction. Nucleotide degradation regulates this process and is mediated through the action of phosphodiesterase (PDE) enzymes, which also play an integral role in a number of disorders including erectile dysfunction [1], asthma [2], and chronic obstructive pulmonary disease [2], as well as schizophrenia, bipolar disorder, and major depression [3]. As research identifies additional potential drug targets, methodologies for measur- ing activity become vital. Figure 1. Phosphate Sensor assay principle. The protein ribbon diagram illustrates the modified We describe here the use and optimization of a simple, flexible reagent phosphate-binding protein with the MDCC fluoro- for measurement of phosphodiesterase activity through coupling it with phore (shown in blue). -
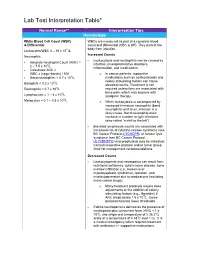
Lab Test Interpretation Table*
Lab Test Interpretation Table* Normal Range** Interpretation Tips Hematology White Blood Cell Count (WBC) WBCs are measured as part of a complete blood & Differential count and differential (CBC & diff). They protect the body from infection. Leukocytes/WBC 4 – 10 x 109 /L Increased Counts Neutrophils - Leukocytosis and neutrophilia can be caused by Absolute Neutrophil Count (ANC) = - infection, myeloproliferative disorders, 2 – 7.5 x 109/L inflammation, and medications. - Calculated ANC = WBC x (segs+bands) / 100 o In cancer patients, supportive - Band neutrophils: < 0.7 x 109/L medications such as corticosteroids and 9 colony stimulating factors can cause Basophils < 0.2 x 10 /L elevated counts. Treatment is not Eosinophils < 0.7 x 109/L required unless they are associated with 9 bone pain, which may improve with Lymphocytes = 1 – 4 x 10 /L analgesic therapy. 9 Monocytes = 0.1 – 0.8 x 10 /L o When leukocytosis is accompanied by increased immature neutrophils (band neutrophils) and fever, infection is a likely cause. Band neutrophils often increase in number to fight infections (also called “a shift to the left”). - Elevated lymphocyte counts are associated with increased risk of cytokine-release syndrome (see BC Cancer Protocol LYCHOPR) or tumour lysis syndrome (see BC Cancer Protocol ULYVENETO) and prophylaxis may be indicated. Consult respective protocol and/or tumor group chair for management recommendations. Decreased Counts - Leukocytopenia and neutropenia can result from nutritional deficiency, autoimmune disease, bone marrow infiltration (i.e., leukemia or myelodysplastic syndrome), radiation, and myelosuppression due to medications (including many cancer drugs). o Many treatment protocols require dose adjustments or the addition of colony stimulating factors (e.g., filgrastim) if ANC drops below 1.5 x 109/L. -

Tissue-Nonspecific Alkaline Phosphatase
biomolecules Review Tissue-Nonspecific Alkaline Phosphatase— A Gatekeeper of Physiological Conditions in Health and a Modulator of Biological Environments in Disease Daniel Liedtke 1,* , Christine Hofmann 2, Franz Jakob 3, Eva Klopocki 1 and Stephanie Graser 3 1 Institute for Human Genetics, Biocenter, Julius-Maximilians-University Würzburg, 97074 Würzburg, Germany; [email protected] 2 Section of Pediatric Rheumatology and Osteology, University Children’s Hospital of Würzburg, 97080 Würzburg, Germany; [email protected] 3 Bernhard-Heine-Center for Locomotion Research, Julius-Maximilians-University Würzburg, 97076 Würzburg, Germany; [email protected] (F.J.); [email protected] (S.G.) * Correspondence: [email protected] Received: 30 October 2020; Accepted: 5 December 2020; Published: 8 December 2020 Abstract: Tissue-nonspecific alkaline phosphatase (TNAP) is a ubiquitously expressed enzyme that is best known for its role during mineralization processes in bones and skeleton. The enzyme metabolizes phosphate compounds like inorganic pyrophosphate and pyridoxal-50-phosphate to provide, among others, inorganic phosphate for the mineralization and transportable vitamin B6 molecules. Patients with inherited loss of function mutations in the ALPL gene and consequently altered TNAP activity are suffering from the rare metabolic disease hypophosphatasia (HPP). This systemic disease is mainly characterized by impaired bone and dental mineralization but may also be accompanied by neurological symptoms, like anxiety disorders, seizures, and depression. HPP characteristically affects all ages and shows a wide range of clinical symptoms and disease severity, which results in the classification into different clinical subtypes. This review describes the molecular function of TNAP during the mineralization of bones and teeth, further discusses the current knowledge on the enzyme’s role in the nervous system and in sensory perception. -

Mechanism of Action of Ribonuclease H Isolated from Avian Myeloblastosis Virus and Escherichia Coli* (RNA-Dependent DNA Polymerase/Processive Exonuclease/E
Proc. Nat. Acad. Sci. USA Vol. 70, No. 2, pp. 466-470, February 1973 Mechanism of Action of Ribonuclease H Isolated from Avian Myeloblastosis Virus and Escherichia coli* (RNA-dependent DNA polymerase/processive exonuclease/E. coli endonuclease) JONATHAN P. LEIS, IRA BERKOWER, AND JERARD HURWITZ Department of Developmental Biology and Cancer, Albert Einstein College of Medicine, Bronx, New York 10461 Communicated by Leon A. Heppel, November 16, 1972 ABSTRACT Purified preparations of RNA-dependent nucleotide kinase (13) were prepared by Drs. R. Silber and DNA polymerase isolated from avian myeloblastosis virus V. G. Malathi. [,y-32P]ATP was purchased from New En- contain RNase H activity. Labeled ribohomopolymers are degraded in the presence of their complementary deoxyri- gland Nuclear; labeled polynucleotides and other nucleotides bopolymer, except [3HJpoly(U).poly(dA). The degradation were from Schwarz/Mann Research or Miles Laboratories, products formed from [3Hpoly(A) . poly(dT) were identified Inc. N-cyclohexyl-N'-3(4-methylmorpholinium)ethyl carbo- as oligonucleotides containing 3'-hydroxyl and 5'-phos- diimide p-toluene sulfonate was from Aldrich Chemical Co. pbate termini, while AMP was not detected. The nuclease has been characterized as a processive exonuclease that All other reagents were described (14). requires ends of poly(A) chains for activity. Exonucleolytic Assay for RNase H. Reaction mixtures (0.05 ml) containing attack occurs in both 5' to 3' and 3' to 5' directions. RNase H has also been purified from E. coli. This nu- 1 4mol of Tris - HCl (pH 8.0), 0.5 umol of MgC12, 0.3 jumol of clease degrades all homoribopolymers tested in the pres- dithioerythritol, 2 ,ug of albumin, 0.52 nmol of poly(dT), ence of their complementary deoxyribopolymers to yield 0.56 nmol of [3H]poly(A), and enzyme were incubated for 30 oligonucleotides with 5'-phosphate and 3'-hydroxyl ter- min at 380. -

A Review of Phosphatidate Phosphatase Assays
REVIEW A review of phosphatidate phosphatase assays Prabuddha Dey1, Gil-Soo Han1 , and George M. Carman1,* 1Department of Food Science and the Rutgers Center for Lipid Research, New Jersey Institute for Food, Nutrition, and Health, Rutgers University, New Brunswick, NJ, USA Abstract Phosphatidate phosphatase (PAP) catalyzes the ganisms studied thus far, PAP activity is dependent on the penultimate step in the synthesis of triacylglycerol and regu- DXDX(T/V) catalytic motif in the haloacid dehalogenase- lates the synthesis of membrane phospholipids. There is like domain (10, 13, 14). The enzyme is extensively modi- much interest in this enzyme because it controls the cellular fied posttranslationally by phosphorylation, which acutely levels of its substrate, phosphatidate (PA), and product, regulates its catalytic activity, subcellular localization, and DAG; defects in the metabolism of these lipid intermediates are the basis for lipid-based diseases such as obesity, lipodys- protein stability (15–17). Downloaded from trophy, and inflammation. The measurement of PAP activity is PAP is a key lipid metabolic enzyme that is required for required for studies aimed at understanding its mechanisms the synthesis of the neutral lipid triacylglycerol (TAG) and of action, how it is regulated, and for screening its activators major membrane phospholipids (16–18) (Fig. 1). The en- and/or inhibitors. Enzyme activity is determined through zyme product, DAG, is a direct precursor of TAG (16, 19–22), the use of radioactive and nonradioactive assays that measure whereas its substrate, PA, is a direct precursor of the lipo- the product, DAG, or Pi. However, sensitivity and ease of use nucleotide, CDP-DAG, a key intermediate in phospholipid www.jlr.org are variable across these methods.