WO 2017/181118 Al 19 October 2017 (19.10.2017) P O P C T
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Drugs Manual September 2019
Drug Enforcement Administration Office of Forensic Sciences Analysis of Drugs Manual September 2019 Date Posted: 10/23/2019 Analysis of Drugs Manual Revision: 4 Issue Date: September 5, 2019 Effective Date: September 9, 2019 Approved By: Nelson A. Santos Table of Contents CHAPTER 1 – QUALITY ASSURANCE ......................................................................... 3 CHAPTER 2 – EVIDENCE ANALYSIS ......................................................................... 93 CHAPTER 3 – FIELD ASSISTANCE .......................................................................... 165 CHAPTER 4 – FINGERPRINT AND SPECIAL PROGRAMS ..................................... 179 Appendix 1A – Definitions ........................................................................................... 202 Appendix 1B – Acronyms and Abbreviations .............................................................. 211 Appendix 1C – Instrument Maintenance Schedule ..................................................... 218 Appendix 1D – Color Test Reagent Preparation and Procedures ............................... 224 Appendix 1E – Crystal and Precipitate Test Reagent Preparation and Procedures .... 241 Appendix 1F – Thin Layer Chromatography................................................................ 250 Appendix 1G – Qualitative Method Modifications ........................................................ 254 Appendix 1H – Analytical Supplies and Services ........................................................ 256 Appendix 2A – Random Sampling Procedures -

Cannabigerol Is a Potential Therapeutic Agent in a Novel Combined Therapy for Glioblastoma
cells Article Cannabigerol Is a Potential Therapeutic Agent in a Novel Combined Therapy for Glioblastoma Tamara T. Lah 1,2,3,*, Metka Novak 1, Milagros A. Pena Almidon 4, Oliviero Marinelli 4 , Barbara Žvar Baškoviˇc 1, Bernarda Majc 1,3, Mateja Mlinar 1, Roman Bošnjak 5, Barbara Breznik 1 , Roby Zomer 6 and Massimo Nabissi 4 1 Department of Genetic Toxicology and Cancer Biology, National Institute of Biology, 1000 Ljubljana, Slovenia; [email protected] (M.N.); [email protected] (B.Ž.B.); [email protected] (B.M.); [email protected] (M.M.); [email protected] (B.B.) 2 Faculty of Chemistry and Chemical Technology, University of Ljubljana, 1000 Ljubljana, Slovenia 3 Jožef Stefan International Postgraduate School, 1000 Ljubljana, Slovenia 4 School of Pharmacy, Experimental Medicine Section, University of Camerino, 62032 Camerino, Italy; [email protected] (M.A.P.A.); [email protected] (O.M.); [email protected] (M.N.) 5 Department of Neurosurgery, University Medical Centre Ljubljana, 1000 Ljubljana, Slovenia; [email protected] 6 MGC Pharmaceuticals d.o.o., 1000 Ljubljana, Slovenia; [email protected] * Correspondence: [email protected]; Tel.: +386-41-651-629 Simple Summary: Among primary brain tumours, glioblastoma is the most aggressive. As early relapses are unavoidable despite standard-of-care treatment, the cannabinoids delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD) alone or in combination have been suggested as a combined treatment strategy for glioblastomas. However, the known psychoactive effects of THC hamper its medical applications in these patients with potential cognitive impairment due to the progression of the Citation: Lah, T.T.; Novak, M.; Pena Almidon, M.A.; Marinelli, O.; disease. -

A Dissertation Entitled Uncovering Cannabinoid Signaling in C. Elegans
A Dissertation Entitled Uncovering Cannabinoid Signaling in C. elegans: A New Platform to Study the Effects of Medicinal Cannabis By Mitchell Duane Oakes Submitted to the Graduate Faculty as partial fulfillment of the requirements for the Doctor of Philosophy Degree in Biology ________________________________________ Dr. Richard Komuniecki, Committee Chair _______________________________________ Dr. Bruce Bamber, Committee Member ________________________________________ Dr. Patricia Komuniecki, Committee Member ________________________________________ Dr. Robert Steven, Committee Member ________________________________________ Dr. Ajith Karunarathne, Committee Member ________________________________________ Dr. Jianyang Du, Committee Member ________________________________________ Dr. Amanda Bryant-Friedrich, Dean College of Graduate Studies The University of Toledo August 2018 Copyright 2018, Mitchell Duane Oakes This document is copyrighted material. Under copyright law, no parts of this document may be reproduced without the expressed permission of the author. An Abstract of Uncovering Cannabinoid Signaling in C. elegans: A New Platform to Study the Effects of Medical Cannabis By Mitchell Duane Oakes Submitted to the Graduate Faculty as partial fulfillment of the requirements for the Doctor of Philosophy Degree in Biology The University of Toledo August 2018 Cannabis or marijuana, a popular recreational drug, alters sensory perception and exerts a range of medicinal benefits. The present study demonstrates that C. elegans exposed to -

Microgram Journal, Vol 2, Number 1
Washington, D. C. Office of Science and Education Vol.II,No.1 Division of Laboratory Operations January 1969 INDEXISSUE CORRECTION 11 "Structure Elucidation of 'LBJ' , by Sander W. Bellman, John W. Turczan, James Heagy and Ted M. Hopes, Micro Gram .!., 3, 6-13 (Dec. 1968) Page 7, third and fourth sentences under Discussion: Change to read: "The melting point of the acid moiety found in step (g) was 148-150°c., compared to the litera ture, v~lue of 151°c for the melting point of benzilic acid (2); thus the benzilic acid melting point gives support to the proposed structure for 'LBJ'. Spectral evidence also supports the proposed structure". MICRO-GRAMREVISION Please re-number the pages of your copies of Micro-Gram, Volume I. Re-number pages bearing printing only. Vol ume I will then be numbered from page 1, the front page of issue No. 1, through page 189 the last page of issue No. 12. To help with this task, pages contained within each issue are as follows: Issue Number Page Through 1 1 8 2 9 29 3 30 32 4 33 66 5 67 79 6 80 97 7 98 120 8 121 128 9 129 136 10 137 157 11 158 170 12 171 189 CAUTION: Use of this publication should be restricted to forensic analysts or others having a legitimate need for this material. From the Archive Library of Erowid Center http://erowid.org/library/periodicals/microgram -2- CANNABIS ,·,-...__/' Attached is a copy of 11A Short Rapid Method for the Identification of Cannabis." The method was developed by Mro H.D. -

Tuning Drug Release from Polyoxazoline-Drug Conjugates T ⁎ J
European Polymer Journal 120 (2019) 109241 Contents lists available at ScienceDirect European Polymer Journal journal homepage: www.elsevier.com/locate/europolj Tuning drug release from polyoxazoline-drug conjugates T ⁎ J. Milton Harrisa, ,1, Michael D. Bentleya, Randall W. Moreaditha, Tacey X. Viegasa,1, Zhihao Fanga, Kunsang Yoona, Rebecca Weimera, Bekir Dizmanb, Lars Nordstiernac a Serina Therapeutics, Inc., 601 Genome Way, Suite 2001, Huntsville, AL 35806, USA2 b Sabanci University, Faculty of Engineering and Natural Sciences, Tuzla, 34956 İstanbul, Turkey2 c Department of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-412 96 Göteborg, Sweden ARTICLE INFO ABSTRACT Keywords: Poly(2-oxazoline)-drug conjugates with drugs attached via releasable linkages are being developed for drug Poly(2-oxazoline) or POZ delivery. Such conjugates with pendent ester linkages that covalently bind drugs to the polymer backbone ex- Poly(2-ethyl-2-oxazoline) or PEOZ hibit significantly slower hydrolytic release rates in plasma than the corresponding PEG- and dextran-drug Pendent drugs conjugates. The slow drug release rates in-vitro of these POZ-drug conjugates contribute to extended in-vivo Degradable ester linkages pharmacokinetic profiles. In some instances, the release kinetics may be relatively sustained and ideal foronce-a- Pharmacokinetics week subcutaneous injection, whereas the native drug by itself may only have an in-vivo half-life of a few hours. Drug delivery Phenolic drugs The origin of this unusual kinetic and pharmacokinetic behavior is proposed here to involve folding of the POZ conjugate such that the relatively hydrophobic drug forms a central core, and the relatively hydrophilic polymer wraps around the core and slows enzymatic attack on the drug-polymer chemical linkage. -

Cannabis, the Endocannabinoid System and Immunity—The Journey from the Bedside to the Bench and Back
International Journal of Molecular Sciences Review Cannabis, the Endocannabinoid System and Immunity—The Journey from the Bedside to the Bench and Back Osnat Almogi-Hazan * and Reuven Or Laboratory of Immunotherapy and Bone Marrow Transplantation, Hadassah Medical Center, The Faculty of Medicine, Hebrew University of Jerusalem, Jerusalem 91120, Israel; [email protected] * Correspondence: [email protected] Received: 21 May 2020; Accepted: 19 June 2020; Published: 23 June 2020 Abstract: The Cannabis plant contains numerous components, including cannabinoids and other active molecules. The phyto-cannabinoid activity is mediated by the endocannabinoid system. Cannabinoids affect the nervous system and play significant roles in the regulation of the immune system. While Cannabis is not yet registered as a drug, the potential of cannabinoid-based medicines for the treatment of various conditions has led many countries to authorize their clinical use. However, the data from basic and medical research dedicated to medical Cannabis is currently limited. A variety of pathological conditions involve dysregulation of the immune system. For example, in cancer, immune surveillance and cancer immuno-editing result in immune tolerance. On the other hand, in autoimmune diseases increased immune activity causes tissue damage. Immuno-modulating therapies can regulate the immune system and therefore the immune-regulatory properties of cannabinoids, suggest their use in the therapy of immune related disorders. In this contemporary review, we discuss the roles of the endocannabinoid system in immunity and explore the emerging data about the effects of cannabinoids on the immune response in different pathologies. In addition, we discuss the complexities of using cannabinoid-based treatments in each of these conditions. -

Annual Report © 2018 Ciberned
2018 ANNUAL REPORT © 2018 CIBERNED Coordination and management of content: Miguel Medina Padilla José de Arriba-Enríquez Aina Frontera Sánchez Almudena Flores Junquera Soledad Valero Rodríguez Design and editorial coordination: OnAccent.com CIBERNED 2018 ANNUAL REPORT INDEX 005 | Letter from the Scientific Director 006 | Introduction 009 | Aims 010 | Directory of research groups and associated institutions 012 | Geographic distribution of CIBERNED research groups 013 | Organizational structure 014 | Organization chart 015 | External scientific advisory committee 016 | Consortium members 017 | Research programmes 019 | Program 1: Alzheimer’s disease and other degenerative dementias 107 | Program 2: Parkinson’s and Huntington’s disease and other degenerative movement disorders 219 | Program 3: Amyotrophic Lateral Sclerosis and other neuromuscular disorders 257 | Cooperative research 279 | International relations 291 | Scientific productivity and other activities 307 | Finantial report 333 | Index of investigators 3 4 CIBERNED 2018 ANNUAL REPORT Jesús Ávila de Grado SCIENTIFIC DIRECTOR LETTER FROM THE SCIENTIFIC DIRECTOR As in previous years, I would like to begin by highlighting the excellent work of the re- search groups that make up CIBERNED and that has allowed us, once again, to be at the forefront of the Centers in biomedical research regarding the quality of publications in high- impact index scientific journals. In order to carry out this work, we have counted on the help and collaboration of the Management Office and the Steering Committee of the Center, as well as its support staff, and the help of the Carlos III Institute of Health and the Associated Institutions. A very important novelty has been the incorporation of six new research groups to CIBERNED, led by Drs. -

The Use of Cannabinoids in Animals and Therapeutic Implications for Veterinary Medicine: a Review
Veterinarni Medicina, 61, 2016 (3): 111–122 Review Article doi: 10.17221/8762-VETMED The use of cannabinoids in animals and therapeutic implications for veterinary medicine: a review L. Landa1, A. Sulcova2, P. Gbelec3 1Faculty of Medicine, Masaryk University, Brno, Czech Republic 2Central European Institute of Technology, Masaryk University, Brno, Czech Republic 3Veterinary Hospital and Ambulance AA Vet, Prague, Czech Republic ABSTRACT: Cannabinoids/medical marijuana and their possible therapeutic use have received increased atten- tion in human medicine during the last years. This increased attention is also an issue for veterinarians because particularly companion animal owners now show an increased interest in the use of these compounds in veteri- nary medicine. This review sets out to comprehensively summarise well known facts concerning properties of cannabinoids, their mechanisms of action, role of cannabinoid receptors and their classification. It outlines the main pharmacological effects of cannabinoids in laboratory rodents and it also discusses examples of possible beneficial use in other animal species (ferrets, cats, dogs, monkeys) that have been reported in the scientific lit- erature. Finally, the article deals with the prospective use of cannabinoids in veterinary medicine. We have not intended to review the topic of cannabinoids in an exhaustive manner; rather, our aim was to provide both the scientific community and clinical veterinarians with a brief, concise and understandable overview of the use of cannabinoids in veterinary -

Pharmacodynamics of Cannabinoids
Open Access Archives of Pharmacy and Pharmaceutical Sciences Review Article Pharmacodynamics of cannabinoids Alexandra Sulcova* ISSN ICCI - International Cannabis and Cannabinoids Institute, Jachymova 26/2, 110 00 Praha, 2639-992X Czech Republic “Pharmacodynamics of cannabinoids “(i.e. a set of biological effects elicited in the *Address for Correspondence: Alexandra Sulcova, M.D, Ph.D, Professor of Pharmacology, living organism by interaction with its biochemical and biophysical functions up to the FCMA, FECNP, FCINP, ICCI - International cellular level) is studied for a long time during both, physiological and pathological Cannabis and Cannabinoids Institute, Jachymova conditions. Cannabinoids received their names according to their natural occurrence 26/2, 110 00 Praha, Czech Republic, Tel: 420 732167678; Email: [email protected] as constituents of Cannabis sativa L. (marijuana). The species was classiied in the “Linnaeus’s Species Plantarum (1753)”, the word “sativa” means things that are Submitted: 12 April 2019 Approved: 07 May 2019 cultivated [1]. For ages, people have used cannabis-based preparations for healing and Published: 08 May 2019 pain suppression until the discovery (in 1897) of aspirin (acetylsalicylic acid) which contemporary medicine uses until today. Chemical investigation of marijuana conirmed Copyright: © 2019 Sulcova A. This is an open access article distributed under the Creative various cannabinoid-type components called cannabinoids (presently estimated at Commons Attribution License, which permits about 150). Regarding their possible pharmacodynamic effects, tetrahydrocannabinol unrestricted use, distribution, and reproduction (THC) and cannabidiol (CBD) are the most explored. The determination of THC structure in any medium, provided the original work is properly cited by means of nuclear magnetic resonance imaging increased sharply the number of professional scientiic reports dealing with the studies of THC pharmacodynamic mechanisms of action [2]. -

Icrs2015 Programme
TH 25 ANNUAL SYMPOSIUM OF THE INTERNATIONAL CANNABINOID RESEARCH SOCIETY WOLFVILLE NOVA SCOTIA JUNE 28 - JULY 3, 2015 TH 25 ANNUAL SYMPOSIUM OF THE INTERNATIONAL CANNABINOID RESEARCH SOCIETY WOLFVILLE JUNE 28 – JULY 3, 2015 Symposium Programming by Cortical Systematics LLC Copyright © 2015 International Cannabinoid Research Society Research Triangle Park, NC USA ISBN: 978-0-9892885-2-1 These abstracts may be cited in the scientific literature as follows: Author(s), Abstract Title (2015) 25th Annual Symposium on the Cannabinoids, International Cannabinoid Research Society, Research Triangle Park, NC, USA, Page #. Funding for this conference was made possible in part by grant 5R13DA016280-13 from the National Institute on Drug Abuse. The views expressed in written conference materials or publications and by speakers and moderators do not necessarily reflect the official policies of the Department of Health and Human Services; nor does mention by trade names, commercial practices, or organizations imply endorsement by the U.S. Government. Sponsors ICRS Government Sponsors National Institute on Drug Abuse Non- Profit Organization Sponsors Kang Tsou Memorial Fund 2015 ICRS Board of Directors Executive Director Cecilia Hillard, Ph.D. President Stephen Alexander, Ph.D. President- Elect Michelle Glass, Ph.D. Past President Ethan Russo, M.D. Secretary Steve Kinsey, Ph.D. Treasurer Mary Abood, Ph.D. International Secretary Roger Pertwee, D . Phil. , D . S c. Student Representative Tiffany Lee, Ph.D. Grant PI Jenny Wiley, Ph.D. Managing Director Jason Schechter, Ph.D. 2015 Symposium on the Cannabinoids Conference Coordinators Steve Alexander, Ph.D. Cecilia Hillard, Ph.D. Mary Lynch, M.D. Jason Schechter, Ph.D. -

NIDA Drug Supply Program Catalog, 25Th Edition
RESEARCH RESOURCES DRUG SUPPLY PROGRAM CATALOG 25TH EDITION MAY 2016 CHEMISTRY AND PHARMACEUTICS BRANCH DIVISION OF THERAPEUTICS AND MEDICAL CONSEQUENCES NATIONAL INSTITUTE ON DRUG ABUSE NATIONAL INSTITUTES OF HEALTH DEPARTMENT OF HEALTH AND HUMAN SERVICES 6001 EXECUTIVE BOULEVARD ROCKVILLE, MARYLAND 20852 160524 On the cover: CPK rendering of nalfurafine. TABLE OF CONTENTS A. Introduction ................................................................................................1 B. NIDA Drug Supply Program (DSP) Ordering Guidelines ..........................3 C. Drug Request Checklist .............................................................................8 D. Sample DEA Order Form 222 ....................................................................9 E. Supply & Analysis of Standard Solutions of Δ9-THC ..............................10 F. Alternate Sources for Peptides ...............................................................11 G. Instructions for Analytical Services .........................................................12 H. X-Ray Diffraction Analysis of Compounds .............................................13 I. Nicotine Research Cigarettes Drug Supply Program .............................16 J. Ordering Guidelines for Nicotine Research Cigarettes (NRCs)..............18 K. Ordering Guidelines for Marijuana and Marijuana Cigarettes ................21 L. Important Addresses, Telephone & Fax Numbers ..................................24 M. Available Drugs, Compounds, and Dosage Forms ..............................25 -
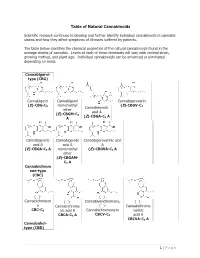
Table of Natural Cannabinoids
Table of Natural Cannabinoids Scientific research continues to develop and further identify individual cannabinoids in cannabis strains and how they affect symptoms of illnesses suffered by patients. The table below identifies the chemical properties of the natural cannabinoids found in the average strains of cannabis. Levels of each of these chemicals will vary with varietal strain, growing method, and plant age. Individual cannabinoids can be enhanced or eliminated depending on need. Cannabigerol- type (CBG) Cannabigerol Cannabigerol Cannabigerovarin (E)-CBG-C monomethyl (E)-CBGV-C 5 Cannabinerolic 3 ether acid A (E)-CBGM-C 5 (Z)-CBGA-C A A 5 Cannabigerolic Cannabigerolic Cannabigerovarinic acid acid A acid A A (E)-CBGA-C5 A monomethyl (E)-CBGVA-C3 A ether (E)-CBGAM- C5 A Cannabichrom ene-type (CBC) (±)- (±)- Cannabichromen (±)- Cannabivarichromene, (±)- e Cannabichrome (±)- Cannabichrome CBC-C5 nic acid A Cannabichromevarin varinic CBCA-C5 A CBCV-C3 acid A CBCVA-C3 A Cannabidiol- type (CBD) 1 | Page (−)-Cannabidiol Cannabidiol Cannabidiol-C4 (−)- Cannabidiorc CBD-C5 momomethyl CBD-C4 Cannabidivarin ol ether CBDV-C3 CBD-C1 CBDM-C5 Cannabidiolic Cannabidivarini acid c acid CBDA-C5 CBDVA-C3 Cannabinodiol- type (CBND) Cannabinodiol Cannabinodivar CBND-C5 in CBND-C3 Tetrahydrocan nabinol-type (THC) 9 9 9 Δ - Δ - Δ - Δ9- Tetrahydrocanna Tetrahydrocan Tetrahydrocannabivarin 9 Tetrahydrocan binol nabinol-C4 Δ -THCV-C3 9 9 nabiorcol Δ -THC-C5 Δ -THC-C4 9 Δ -THCO-C1 9 9 Δ -Tetrahydro- Δ9-Tetrahydro- Δ -Tetrahydro- Δ9-Tetrahydro- cannabinolic