Process for Producing 6-Methyl-2-Heptanone Analogues, and Process for Producing Phyton and Isophytol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Retention Indices for Frequently Reported Compounds of Plant Essential Oils
Retention Indices for Frequently Reported Compounds of Plant Essential Oils V. I. Babushok,a) P. J. Linstrom, and I. G. Zenkevichb) National Institute of Standards and Technology, Gaithersburg, Maryland 20899, USA (Received 1 August 2011; accepted 27 September 2011; published online 29 November 2011) Gas chromatographic retention indices were evaluated for 505 frequently reported plant essential oil components using a large retention index database. Retention data are presented for three types of commonly used stationary phases: dimethyl silicone (nonpolar), dimethyl sili- cone with 5% phenyl groups (slightly polar), and polyethylene glycol (polar) stationary phases. The evaluations are based on the treatment of multiple measurements with the number of data records ranging from about 5 to 800 per compound. Data analysis was limited to temperature programmed conditions. The data reported include the average and median values of retention index with standard deviations and confidence intervals. VC 2011 by the U.S. Secretary of Commerce on behalf of the United States. All rights reserved. [doi:10.1063/1.3653552] Key words: essential oils; gas chromatography; Kova´ts indices; linear indices; retention indices; identification; flavor; olfaction. CONTENTS 1. Introduction The practical applications of plant essential oils are very 1. Introduction................................ 1 diverse. They are used for the production of food, drugs, per- fumes, aromatherapy, and many other applications.1–4 The 2. Retention Indices ........................... 2 need for identification of essential oil components ranges 3. Retention Data Presentation and Discussion . 2 from product quality control to basic research. The identifi- 4. Summary.................................. 45 cation of unknown compounds remains a complex problem, in spite of great progress made in analytical techniques over 5. -

VITAMIN K1 | C31H46O2 - Pubchem
VITAMIN K1 | C31H46O2 - PubChem https://pubchem.ncbi.nlm.nih.gov/compound/Phylloquinone#secti... NIH U.S. National Library of Medicine National Center for Biotechnology Information OPEN CHEMISTRY Search Compounds ! DATABASE VITAMIN K1 " Cite this Record # $ % & ' ( STRUCTURE VENDORS DRUG INFO PHARMACOLOGY LITERATURE PATENTS BIOACTIVITIES PubChem CID: 5284607 VITAMIN K1; Phytonadione; Phylloquinone; 84-80-0; Phytylmenadione; Chemical Names: Phyllochinon More... Molecular Formula: C31H46O2 Molecular Weight: 450.707 g/mol InChI Key: MBWXNTAXLNYFJB-NKFFZRIASA-N Drug Indication Therapeutic Uses Clinical Trials FDA Orange Book Drug Information: FDA UNII Safety Summary: Laboratory Chemical Safety Summary (LCSS) VITAMIN K1 is a family of phylloquinones that contains a ring of 2-methyl-1,4-naphthoquinone and an isoprenoid side chain. Members of this group of vitamin K 1 have only one double bond on the proximal isoprene unit. Rich sources of vitamin K 1 include green plants, algae, and photosynthetic bacteria. Vitamin K1 has antihemorrhagic and prothrombogenic activity. " from MeSH Vitamin K is a family of fat-soluble compounds with a common chemical structure based on 2-methyl-1, 4-naphthoquinone " Metabolite Description from Human Metabolome Database (HMDB) PUBCHEM ) COMPOUND ) VITAMIN K1 Modify Date: 2018-01-06; Create Date: 2004-09-16 1 di 57 12/01/18, 11:36 VITAMIN K1 | C31H46O2 - PubChem https://pubchem.ncbi.nlm.nih.gov/compound/Phylloquinone#secti... * Contents 1 2D Structure 2 3D Conformer 3 Names and Identifiers + 4 Chemical and Physical Properties 5 Related Records 6 Chemical Vendors 7 Drug and Medication Information 8 Pharmacology and Biochemistry 9 Use and Manufacturing 10 Identification 11 Safety and Hazards 12 Toxicity 13 Literature 14 Patents 15 Biomolecular Interactions and Pathways 16 Biological Test Results 17 Classification 18 Information Sources 2 di 57 12/01/18, 11:36 VITAMIN K1 | C31H46O2 - PubChem https://pubchem.ncbi.nlm.nih.gov/compound/Phylloquinone#secti.. -

Vitamin E: Food Chemistry, Composition, and Analysis, Ronald Eitenmiller and Junsoo Lee
Vitamin E Copyright © 2004 by Marcel Dekker, Inc. FOOD SCIENCE AND TECHNOLOGY A Series of Monographs, Textbooks, and Reference Books EDITORIAL BOARD Senior Editors Owen R.Fennema University of Wisconsin-Madison Y.H.Hui Science Technology System Marcus Karel Rutgers University (emeritus) Pieter Walstra Wageningen University John R.Whitaker University of California-Davis Additives P.Michael Davidson University of Tennessee-Knoxville Dairy science James L.Steele University of Wisconsin-Madison Flavor chemistry and sensory analysis John H.Thorngate III University of California-Davis Food engineering Daryl B.Lund University of Wisconsin-Madison Food lipids and flavors David B.Min Ohio State University Food proteins/food chemistry Rickey Y.Yada University of Guelph Health and disease Seppo Salminen University of Turku, Finland Nutrition and nutraceuticals Mark Dreher Mead Johnson Nutritionals Phase transition/food microstructure Richard W.Hartel University of Wisconsin-Madison Processing and preservation Gustavo V.Barbosa-Cánovas Washington State University-Pullman Safety and toxicology Sanford Miller University of Texas-Austin 1. Flavor Research: Principles and Techniques, R.Teranishi, I.Hornstein, P.Issenberg, and E.L.Wick 2. Principles of Enzymology for the Food Sciences, John R.Whitaker 3. Low-Temperature Preservation of Foods and Living Matter, Owen R. Fennema, William D.Powrie, and Elmer H.Marth 4. Principles of Food Science Part I: Food Chemistry, edited by Owen R.Fennema Part II: Physical Principles of Food Preservation, Marcus Karel, Owen R.Fennema, and Daryl B.Lund 5. Food Emulsions, edited by Stig E.Friberg 6. Nutritional and Safety Aspects of Food Processing, edited by Steven R.Tannenbaum 7. Flavor Research: Recent Advances, edited by R.Teranishi, Robert A. -

Chemistry of the Main Component of Essential Oil of Litsea Cubeba and Its Derivatives
Open Journal of Forestry, 2014, 4, 457-466 Published Online October 2014 in SciRes. http://www.scirp.org/journal/ojf http://dx.doi.org/10.4236/ojf.2014.45050 Chemistry of the Main Component of Essential Oil of Litsea cubeba and Its Derivatives Lisong Hu, Menghao Du, Jingping Zhang, Yangdong Wang Research Institute of Subtropical Forestry Chinese Academy of Forestry, Fuyang, China Email: [email protected] Received 8 July 2014; revised 11 August 2014; accepted 26 August 2014 Copyright © 2014 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract The tree of Litsea cubeba is widely spread in China, Indonesia and other part of Southeast Asia. The essential oil of Litsea cubeba (EOLC) is obtained by steam distillation from the pepper-like fruits tree Litsea cubeba. The EOLC consists of about 29 active compounds. Among them, citral is the main component; the content of citral is nearly 80% of the EOLC. Due to the special function group, citral is easy to react with many chemicals. Thus, EOLC is usually applied as starting ma- terial to carry out aldol condensation, reduction, and six-member ring forming reaction. The EOLC is extensively employed to synthesis of geranal nitriles, pseudonoe, ionone, methyl ionone, Vita- min E and Vitamin A. These products are broadly applied in the fields of fragrance, perfume, med- icine and so on. This paper presents comprehensive utilization of EOLC as raw materials to syn- thesize many active chemicals. Keywords Litsea cubeba, Essential Oil, Citral, Synthesis 1. -

Anne Marie Api, Ph.D
Anne Marie Api, Ph.D. W: +1-201-689-8089 ext. 103 C: +1-914-433-1205 Summary of Experience Research Institute for Fragrance Materials Inc. (RIFM) 1984-present Woodcliff Lake, New Jersey USA Vice President, Human Health Sciences 2006-present RIFM, the most comprehensive source worldwide for toxicology data, literature and information on the evaluation of fragrance materials is the international scientific authority for the safe use of fragrance materials. Combining an advanced knowledge of fragrance ingredient safety, senior leadership experience, an excellent work ethic and exceptional communication skills, I have established a quality record of managing fragrance ingredient safety at RIFM. I am committed to investigating new scientific methodologies to keep RIFM at the cutting edge of advancements in toxicology. I am also committed to education and inspiring others on current and future goals. Responsible for the human health scientific program. Continue to maintain and oversee implementation of the RIFM Safety Assessment program with respect to all human health endpoints as well as computational toxicology. Investigate and initiate new research and testing projects, including those that would involve development of new testing methodologies. I continue to promote and initiate new research projects that evaluate and improve test methodologies and assessment procedures to address the emerging safety challenges. Initiated and oversee the publication of the RIFM safety assessments on the Food and Chemical Toxicology Fragrance Material Safety Assessment Center (http://fragrancematerialsafetyresource.elsevier.com). This is a partnership between RIFM and Elsevier for an open access resource center. Initiated and continue to update an aggregate exposure model for providing a realistic exposure assessment to fragrance ingredients. -

US5236950.Pdf
|||||||||||||||| USOO.5236950A United States Patent (19) 11 Patent Number: 5,236,950 Aoyama et al. 45 Date of Patent: Aug. 17, 1993 (54) PROCESS FOR HAIR GROWTH 2652256 6/1977 Fed. Rep. of Germany . 2812978 10/1979 Fed. Rep. of Germany . 75) Inventors: Hajime Aoyama; Satoshi Ono; Osamu 3738405 5/1989 Fed. Rep. of Germany . Oohashi; Hirokazu Narita; Shuntaro 119.0002 10/1959 France, Takano, all of Toyama, Japan 60-004113 10/1985 Japan. 61-207321 9/1986 Japan . 73) Assignee: Toyama Chemical Co., Ltd., Tokyo, 8302390 7/1983 PCT Int'l Appl. Japan 780801 8/1957 United Kingdom . 923400 4/1963 United Kingdom . 21 Appl. No.: 311,945 944834 12/1963 United Kingdom. (22 Filed: Feb. 17, 1989 83/02390 7/1983 World int. Prop. O. (30) Foreign Application Priority Data OTHER PUBLICATIONS Feb. 18, 1988 JP Japan .................................. 63-33968 Derwent Abstract WPI Acc No. 75-62757W/38 Jun. 3, 1988 JP Japan ................................ 63.36824 (Takasago). 51) Int. Cl................................................. A61K 7/06 Derwent Abstract WPI Acc No. JP49069845 52 U.S. Cl. .................................... 514/478; 514/354; (Takasago). 514/356; 514/532; 514/556; 514/715; 514/738; Derwent-Ref: 60454Y/34 Nov. 1, 1976. 514/739 Derwent-Ref: 85-077293/13 Jul. 25, 1983. 58) Field of Search ................. 514/70,478, 556, 354, Derwent-Ref: 85-077294/13 Jul. 28, 1983. 514/356, 532, 715, 738 Derwent-Ref: 86-141951/22 Sep. 27, 1984. Derwent-Ref: 62505B/34 Dec. 27, 1977. (56) References Cited Chemical Abstracts, vol. 83, p. 435, 1975, No. 65330r, U.S. PATENT DOCUMENTS Yoshigi Hideki, et al., "Cosmetics With Improved 4,139,619 2/1979 Chidsey, III et al. -

Acetylene in Organic Synthesis: Recent Progress and New Uses
Review Acetylene in Organic Synthesis: Recent Progress and New Uses Vladimir V. Voronin 1, Maria S. Ledovskaya 1, Alexander S. Bogachenkov 1, Konstantin S. Rodygin 1 and Valentine P. Ananikov 1,2,* 1 Institute of Chemistry, Saint Petersburg State University, Universitetsky prospect 26, Peterhof 198504, Russia; [email protected] (V.V.V.); [email protected] (M.S.L.); [email protected] (A.S.B.); [email protected] (K.S.R.) 2 N. D. Zelinsky Institute of Organic Chemistry Russian Academy of Sciences, Leninsky prospect 47, Moscow 119991, Russia * Correspondence: [email protected] Received: 16 August 2018; Accepted: 17 September 2018; Published: 24 September 2018 Abstract: Recent progress in the leading synthetic applications of acetylene is discussed from the prospect of rapid development and novel opportunities. A diversity of reactions involving the acetylene molecule to carry out vinylation processes, cross-coupling reactions, synthesis of substituted alkynes, preparation of heterocycles and the construction of a number of functionalized molecules with different levels of molecular complexity were recently studied. Of particular importance is the utilization of acetylene in the synthesis of pharmaceutical substances and drugs. The increasing interest in acetylene and its involvement in organic transformations highlights a fascinating renaissance of this simplest alkyne molecule. Keywords: acetylene; vinylation; cross-coupling; addition reactions; drugs; pharmaceutical substances; biologically active molecule; monomers; polymers 1. Introduction Since the discovery of acetylene, new areas of acetylene chemistry have been continuously developed. The rich scope of chemical transformations available for a C≡C triple bond can be exemplified by coupling [1–5] and addition reactions [6,7]. -

Butanol Production from Lignocellulosic Feedstocks by Acetone-Butanol-Ethanol Fermentation with Integrated Product Recovery
Butanol Production from Lignocellulosic Feedstocks by Acetone-Butanol-Ethanol Fermentation with Integrated Product Recovery Dissertation Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduation School of The Ohio State University By Congcong Lu, M.S. Graduate Program in Chemical and Biomolecular Engineering The Ohio State University 2011 Dissertation Committee: Professor Shang-Tian Yang, Advisor Professor Jeffrey Chalmers Professor Andre Palmer 1 Copyright by Congcong Lu 2011 2 Abstract n-Butanol has been attracting research attention as a liquid biofuel recently, in addition to its current application as an industrial chemical and solvent. With the concerns of diminishing fossil reserves, environmental issues caused by greenhouse gas emission, and unstable supply and price spike of crude oil, renewed interests have returned to pursue biobutanol production through acetone-butanol-ethanol (ABE) fermentation as opposed to petrochemically-derived butanol. However, the conventional ABE fermentation suffers from many limitations, including low butanol titer, high cost of traditional food-based raw materials, end-product inhibition and high butanol recovery cost by distillation, which negatively impacts the process efficiency and economics. Fortunately, these hurdles are being overcome by technological advances on ABE fermentation in the past few decades. Research on genetic modifications and chemical mutation of solventogenic Clostridia has focused on obtaining mutant strains with enhanced butanol producing ability. Adequate research success in utilizing renewable and sustainable lignocellulosic biomass has identified a novel group of cost-effective feedstocks for ABE fermentation in replacement of the traditional costly starch and sugar-based substrates. Novel fed-batch ii and continuous fermentation processes with cell immobilization and cell recycle have been developed for more efficient substrate conversion and butanol production. -
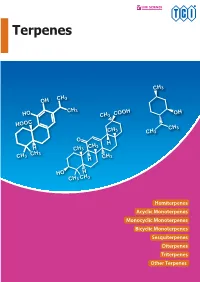
Please Inquire for Pricing and Availability of Listed Products to Our Local Sales Representatives
Please inquire for pricing and availability of listed products to our local sales representatives. 1 Terpenes Terpenes are a large family of natural products and are known ● Solubility to be the primary constituents of essential oils. They are In general, most of the terpenes are insoluble in water but soluble biosynthesized via the mevalonate pathway. The basic structure in ethanol, chloroform and diethyl ether. They can be added to the is derived from five-carbon isoprene units1,2) which are linked buffer solution as a dimethyl sulfoxide solution to examine their together in a head-to-tail fashion to form linear chains or rings. activity in living organisms. Please take caution that the solution They can be classified on the basis of the length of the carbon becomes suspended as the concentration level of the dissolved chains as illustrated below. substance increases. It is recommended to define the optimal concentration level and volume of addition in advance. Glycosides of terpenes are more water-soluble than their aglycones. Table 1. Classification of Terpenes and xamples Number Name of xamples Carbons Hemiterpenes 5 Isoprene ● Stability Monoterpenes 1 Menthol, Geraniol: Flavors, Food Additives In general, monoterpenes are relatively stable. However, oily Artemisinin: Antimalarial Drug Sesquiterpenes 15 α-Bisaborol: Flavor, Cosmetic Ingredients sesquiterpenes and diterpenes are less stable bearing more Diterpenes 2 Paclitaxel: Antitumor Agent, Gibberellins: Plant Hormones oxygen fuctional groups rendering them unsuitable for storage Triterpenes 3 Lanosterol: Precursor of Steroid Biosynthesis over longer periods. However, most of the triterpenes are solid and show good stability. Hemiterpenes(C5) ● Detection Steroids (C18, 19, 21, 24, 27) Monoterpenes (C10 ) Since some terpenes do not have chromophore unit, UV detection by normal-phase HPLC is difficult. -

Industrial Chemicals, Ashfords Dictionary, Chemical Name Index
2 Chemical name index A A ACA 4-AA ACA AA ACAC AA acamprosate calcium AAA acarbose AABA ACB AADMC 7-ACCA AAEM ACE AAMX acebutolol AAOA aceclidine AAOC aceclofenac AAOT acemetacin AAPP acenaphthene AAPT acenocoumarol 4-ABA acephate ABA acepromazine abacavir acequinocyl ABAH acesulfame-K abamectin acetaldehyde ABFA acetaldehyde ethyl phenethyl diacetal ABL acetaldehyde oxime ABPA acetaldehyde n-propyl phenethyl diacetal ABS acetaldoxime ABS acetal resins O-ABTF acetal resins P-ABTF acetamide ACN acetamidine hydrochloride 7-ACA 5-acetamido-2-aminobenzenesulfonic acid 7-ANCA 6-acetamido-2-aminophenol-4-sulfonic acid Name Index: Ashford’s Dictionary of Industrial Chemicals 3-acetamidoaniline 3-acetamidoaniline acetic acid, s-butyl ester 4-acetamidoaniline acetic acid, calcium salt p-acetamidoanisole acetic acid, chromium salt 4-acetamidobenzenesulfonyl chloride acetic acid, cinnamyl ester 2-acetamidocinnamic acid acetic acid, citronellyl ester 3-acetamido-2-hydroxyaniline-5-sulfonic acid acetic acid, cobalt salt 1-acetamido-7-hydroxynaphthalene acetic acid, decahydro-β-naphthyl ester 8-acetamido-1-hydroxynaphthalene-3,6-disulfonic acid acetic acid, dicyclopentenyl ester 1-acetamido-7-naphthol acetic acid, diethylene glycol monobutyl ether ester 4-acetamidonitrobenzene acetic acid, 2-ethoxyethyl ester p-acetamidophenol acetic acid, ethoxypropyl ester 3-acetamidopropylsulfonic acid, calcium salt acetic acid, ethyl diglycol ester 5-acetamido-2,4,6-triiodo-N-methylisophthalamic acid acetic acid, ethylene glycol diester 4-acetamido-2-ethoxybenzoic -

RIFM Fragrance Ingredient Safety Assessment, 3,7-Dimethyl-1,6- Nonadien-3-Ol, CAS Registry Number 10339-55-6
Food and Chemical Toxicology 97 (2016) S168eS179 Contents lists available at ScienceDirect Food and Chemical Toxicology journal homepage: www.elsevier.com/locate/foodchemtox Short review RIFM fragrance ingredient safety assessment, 3,7-dimethyl-1,6- nonadien-3-ol, CAS Registry Number 10339-55-6 * A.M. Api a, , D. Belsito b, S. Bhatia a, M. Bruze c, P. Calow d, M.L. Dagli e, W. Dekant f, A.D. Fryer g, L. Kromidas a,S.LaCavaa, J.F. Lalko a, A. Lapczynski a, D.C. Liebler h, Y. Miyachi i, V.T. Politano a, G. Ritacco a, D. Salvito a, T.W. Schultz j, J. Shen a, I.G. Sipes k, B. Wall a, D.K. Wilcox a a Research Institute for Fragrance Materials, Inc., 50 Tice Boulevard, Woodcliff Lake, NJ 07677, USA b Member RIFM Expert Panel, Columbia University Medical Center, Department of Dermatology, 161 Fort Washington Ave., New York, NY 10032, USA c Member RIFM Expert Panel, Malmo University Hospital, Department of Occupational & Environmental Dermatology, Sodra Forstadsgatan 101, Entrance 47, Malmo SE 20502, Sweden d Member RIFM Expert Panel, University of Nebraska Lincoln, 230 Whittier Research Center, Lincoln NE 68583-0857, USA e Member RIFM Expert Panel, University of Sao Paulo, School of Veterinary Medicine and Animal Science, Department of Pathology, Av. Prof. dr. Orlando Marques de Paiva, 87, Sao Paulo CEP 05508-900, Brazil f Member RIFM Expert Panel, University of Wuerzburg, Department of Toxicology, Versbacher Str. 9, 97078 Würzburg, Germany g Member RIFM Expert Panel, Oregon Health Science University, 3181 SW Sam Jackson Park Rd., Portland, -
Process for Producing 6-Methyl-3-Hepten-2-One and 6-Methyl-2-Heptanone Analogues, and Process for Producing Phyton Or Isophytol
* — llll INI II III II III IN II II II I II OJII Eur°Pean Patent Office <*S Office europeen des brevets (11) EP0 816 321 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int. CI.6: C07C 45/74, C07C 45/62, 07.01.1998 Bulletin 1998/02 C07C 45/73 C07C 49/04, (21) Application number: 97111203.2 C07C 49/203, C07C 33/03 (22) Date of filing: 03.07.1997 (84) Designated Contracting States: • Kumagai, Noriaki AT BE CH DE DK ES Fl FR GB GR IE IT LI LU MC Kashima-gun, Ibaraki, 314-02 (JP) NL PT SE • Iwasaki, Hideharu Kashima-gun, Ibaraki, 314-02 (JP) (30) Priority: 05.07.1996 JP 195480/96 • Onishi, Takashi 11.12.1996 JP 352214/96 Kashima-gun, Ibaraki, 314-02 (JP) 06.03.1997 JP 51356/97 • Ueyama, Fuyuo Kitakanbara-gun, Niigata, 959-26 (JP) (71) Applicant: KURARAY Co. LTD. Okayama710(JP) (74) Representative: Muller-Bore & Partner (72) Inventors: Patentanwalte • Kido, Yoichi Grafinger Strasse 2 Kashima-gun, Ibaraki, 314-02 (JP) 81671 Munchen (DE) (54) Process for producing 6-methyl-3-hepten-2-one and 6-methyl-2-heptanone analogues, and process for producing phyton or isophytol (57) Provided are a process for producing 6-methyl-3-hepten-2-one by cross aldol condensation carried out while each continuously adding to acetone, isovaleraldehyde and an aqueous alkali containing an alkaline substance; a proc- ess for producing a 6-methyl-2-heptanone analogue represented by Formula (1): (1) wherein n is an integer of 0 or 1 or more; which comprises allowing hydrogen, acetone and an aldehyde represented by Formula (2): (2) ^ wherein n is as defined above; X and Y each represents a hydrogen atom or they are coupled together to form a carbon- .,— carbon bond; and Z and W each represents a hydrogen atom or they are coupled together to form a carbon-carbon CM bond; ^ to react in the presence of an aqueous alkali containing an alkaline substance, and a hydrogenation catalyst; and a process for producing phyton or isophytol using the 6-methyl-3-hepten-2-one or the 6-methyl-2-heptanone analogue.