Chapter 4 HEMOSTASIS and BLOOD
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Point-Of-Care Testing for Anticoagulation Monitoring In
Point-of-Care Testing for Anticoagulation PATIENT SAFETY Monitoring in Neuroendovascular Procedures H.M. Hussein SUMMARY: POC testing is defined as diagnostic testing at or near the site of patient care. Rapid A.L. Georgiadis measurement of the intensity of anticoagulation and, more recently, platelet inhibition allows dose titration of adjuvant medications such a heparin and antiplatelet agents during neuroendovascular A.I. Qureshi procedures. However, knowledge among practicing physicians regarding the pathophysiologic basis of these measurements and variations in knowledge about the differences among devices is often limited. This review discusses the role of anticoagulation in endovascular procedures and the currently available POC tests for anticoagulation monitoring. ABBREVIATIONS: ACT ϭ activated clotting time; Anti-Xa ϭ quantitative chromogenic heparin assay; aPTT ϭ activated partial thromboplastin time; AT ϭ antithrombin; CI ϭ confidence interval; GP IIb/IIIa ϭ glycoprotein IIb/IIIa; Hemonox-CT ϭ Hemonox clotting time; HIT ϭ heparin-induced thrombocytopenia; HMT ϭ Heparin Management Test; IV ϭ intravenous; LMWH ϭ low-molecular- weight heparin; MI ϭ myocardial infarction; OR ϭ odds ratio; PCI ϭ percutaneous coronary intervention; POC ϭ point of care; UFH ϭ unfractionated heparin hromboembolism ensues from activation of platelets and Brief Overview of Anticoagulant Medications Tthe coagulation cascade and is a major source of compli- Anticoagulants reduce the activity of proteases in the coagula- cations during endovascular procedures. Multiple studies tion cascade. On the basis of their mechanism of action (Fig 1), have demonstrated accelerated platelet activation during cor- anticoagulants can be divided into 4 major groups: vitamin K onary and cerebral angioplasty.1,2 The coagulation cascade antagonists (warfarin), heparins (UFH and LMWH), direct (Fig 1) consists of a sequential conversion of a series of proen- thrombin inhibitors, and direct factor Xa inhibitors. -

Section 8: Hematology CHAPTER 47: ANEMIA
Section 8: Hematology CHAPTER 47: ANEMIA Q.1. A 56-year-old man presents with symptoms of severe dyspnea on exertion and fatigue. His laboratory values are as follows: Hemoglobin 6.0 g/dL (normal: 12–15 g/dL) Hematocrit 18% (normal: 36%–46%) RBC count 2 million/L (normal: 4–5.2 million/L) Reticulocyte count 3% (normal: 0.5%–1.5%) Which of the following caused this man’s anemia? A. Decreased red cell production B. Increased red cell destruction C. Acute blood loss (hemorrhage) D. There is insufficient information to make a determination Answer: A. This man presents with anemia and an elevated reticulocyte count which seems to suggest a hemolytic process. His reticulocyte count, however, has not been corrected for the degree of anemia he displays. This can be done by calculating his corrected reticulocyte count ([3% × (18%/45%)] = 1.2%), which is less than 2 and thus suggestive of a hypoproliferative process (decreased red cell production). Q.2. A 25-year-old man with pancytopenia undergoes bone marrow aspiration and biopsy, which reveals profound hypocellularity and virtual absence of hematopoietic cells. Cytogenetic analysis of the bone marrow does not reveal any abnormalities. Despite red blood cell and platelet transfusions, his pancytopenia worsens. Histocompatibility testing of his only sister fails to reveal a match. What would be the most appropriate course of therapy? A. Antithymocyte globulin, cyclosporine, and prednisone B. Prednisone alone C. Supportive therapy with chronic blood and platelet transfusions only D. Methotrexate and prednisone E. Bone marrow transplant Answer: A. Although supportive care with transfusions is necessary for treating this patient with aplastic anemia, most cases are not self-limited. -
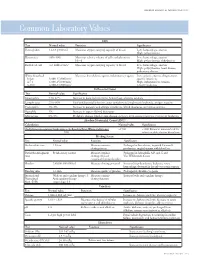
Common Laboratory Values
AmericAn AcAdemy of PediAtric dentistry Reference Manual 2006-2007 Resource Section 251 Common LaboratoryCommon Values Laboratory Values CBC Test Normal value Function Significance Hemoglobin 12-18 g/100 mL Measures oxygen carrying capacity of blood Low: hemorrhage, anemia High: polycythemia Hematocrit 35%-50% Measures relative volume of cells and plasma in Low: hemorrhage, anemia blood High: polycythemia, dehydration Red blood cell 4-6 million/mm3 Measures oxygen-carrying capacity of blood Low: hemorrhage, anemia High: polycythemia, heart disease, pulmonary disease White blood cell Measures host defense against inflammatory agents Low: aplastic anemia, drug toxicity, Infant 8,000-15,000/mm3 specific infections 4-7 y 6,000-15,000/mm3 High: inflammation, trauma, 8-18 y 4,500-13,500/mm3 toxicity, leukemia Differential Count Test Normal value Significance Neutrophils 54%-62% Increase in bacterial infections, hemorrhage, diabetic acidosis Lymphocytes 25%-30% Viral and bacterial infection, acute and chronic lymphocytic leukemia, antigen reaction Eosinophils 1%-3% Increase in parasitic and allergic conditions, blood dyscrasias, pernicious anemia Basophils 1% Increase in types of blood dyscrasias Monocytes 0%-9% Hodgkin’s disease, lipid storage disease, recovery from severe infections, monocytic leukemia Absolute Neutrophil Count (ANC) Calculation Normal value Significance (% Polymorphonuclear Leukocytes + % Bands)×Total White Cell Count >1500 <1000 Patient at increased risk for 100 infection; defer elective dental care Bleeding Screen Test -

Prolonged Partial Thromboplastin Time Without Bleeding History; Fletcher Factor Deficiency
Prolonged Partial Thromboplastin Time Without Bleeding History; Fletcher Factor Deficiency Celalettin ÜSTÜN*, Anand JILLELLA*, Linda HENDRIKS*, Mary JONAH**, Ferdane KUTLAR*, Russell BURGESS*, Abdullah KUTLAR* * Section of Hematology Oncology, Department of Medicine, Medical College of Georgia, ** Department of Pathology, Medical College of Georgia, Augusta, USA ABSTRACT A 67-year-old patient was admitted to the hospital to perform an esophagogastrectomy because a lesion at the lower esophagus was strongly suspicious for cancer. Her medical history and her family his- tory were negative for bleeding tendency or thrombosis. Her activated partial thromboplastin time (aPTT) was prolonged (44 s) whereas her prothrombin time (PT) was normal (11 s) presurgery. Mixing of her plasma with normal plasma corrected her prolonged aPTT (27.9 s). Prolonged incubation shortened the patient’s aPTT (36.3 s). Fletcher factor activity was found to be 50%. The patient underwent an esopha- gogastrectomy without bleeding complications under spinal anesthesia. Fletcher factor deficiency, a ra- re disorder, should be considered in patients who have no history of bleeding tendency with a prolonged aPTT. Surgical interventions are safe in these patients. Key Words: aPTT, Surgery, Fletcher factor, Prekallikrein. Turk J Haematol 2002;19(3): 417-419 Received: 20.06.2001 Accepted: 28.07.2001 INTRODUCTION unt[2]. Hematologists are frequently consulted for preoperative coagulation abnormalities. Preoperative evaluation of hemostasis is cru- cial to assess the risk of per-and peri-operative Prolonged aPTT is not an uncommon abnor- bleeding. The most effective screening method is mality encountered during preoperative evaluati- to obtain a thorough history of bleeding[1]. on. It may indicate the presence of antiphospholi- Preoperative screening tests mostly include acti- pid antibodies or a factor deficiency in the intrinsic vated partial thromboplastin time (aPTT), proth- and/or common pathways of blood coagulation. -

Haptoglobin and Its Related Protein, Zonulin—What Is Their Role in Spondyloarthropathy?
Journal of Clinical Medicine Review Haptoglobin and Its Related Protein, Zonulin—What Is Their Role in Spondyloarthropathy? Magdalena Chmieli ´nska 1,2,* , Marzena Olesi ´nska 2, Katarzyna Romanowska-Próchnicka 1,2 and Dariusz Szukiewicz 1 1 Department of Biophysics and Human Physiology, Medical University of Warsaw, Chałubi´nskiego5, 02-004 Warsaw, Poland; [email protected] (K.R.-P.); [email protected] (D.S.) 2 Department of Connective Tissue Diseases, National Institute of Geriatrics, Rheumatology and Rehabilitation, Sparta´nska1, 02-637 Warsaw, Poland; [email protected] * Correspondence: [email protected] Abstract: Haptoglobin (Hp) is an acute phase protein which supports the immune response and protects tissues from free radicals. Its concentration correlates with disease activity in spondy- loarthropathies (SpAs). The Hp polymorphism determines the functional differences between Hp1 and Hp2 protein products. The role of the Hp polymorphism has been demonstrated in many diseases. In particular, the Hp 2-2 phenotype has been associated with the unfavorable course of some inflammatory and autoimmune disorders. Its potential role in modulating the immune system in SpA is still unknown. This article contains pathophysiological considerations on the potential relationship between Hp, its polymorphism and SpA. Keywords: haptoglobin polymorphism; inflammation; pathogenesis; spondyloarthropathy; zonulin Citation: Chmieli´nska,M.; Olesi´nska, M.; Romanowska-Próchnicka, K.; Szukiewicz, D. Haptoglobin and Its 1. Introduction Related Protein, Zonulin—What Is Spondyloarthropathy is one of the most common rheumatic diseases whose prevalence Their Role in Spondyloarthropathy? J. varies between 0.4 and 1.9% in different countries [1]. The heterogeneity of SpA is the Clin. Med. 2021, 10, 1131. -

Downloaded from Bioscientifica.Com at 09/25/2021 07:25:24AM Via Free Access 812 M Andreassen and Others EUROPEAN JOURNAL of ENDOCRINOLOGY (2012) 166
European Journal of Endocrinology (2012) 166 811–819 ISSN 0804-4643 CLINICAL STUDY GH activity and markers of inflammation: a crossover study in healthy volunteers treated with GH and a GH receptor antagonist Mikkel Andreassen1, Jan Frystyk2,3, Jens Faber1,4 and Lars Østergaard Kristensen1 1Endocrine Unit, Laboratory of Endocrinology 54o4, Department of Internal Medicine O, Herlev Hospital, University of Copenhagen, Herlev Ringvej 75, DK-2730 Herlev, Denmark, 2Department of Endocrinology and Internal Medicine, Aarhus University Hospital, Aarhus, Denmark and 3Medical Research Laboratories, Faculty of Health Sciences, Institute of Clinical Medicine, Aarhus University, Aarhus, Denmark and 4Faculty of Health Science, Copenhagen University, Copenhagen, Denmark (Correspondence should be addressed to M Andreassen; Email: [email protected]) Abstract Introduction: The GH/IGF1 axis may modulate inflammatory processes. However, the relationship seems complicated as both pro- and anti-inflammatory effects have been demonstrated. Methods/design: Twelve healthy volunteers (mean age 36, range 27–49 years) were treated in random order with increasing doses of GH for 3 weeks (first week 0.01 mg/kg per day, second week 0.02 mg/kg per day, and third week 0.03 mg/kg per day) or a GH receptor antagonist (pegvisomant; first week 10 mg/day and last two weeks 15 mg/day), separated by 8 weeks of washout. Circulating levels of the pro-inflammatory cytokines tumor necrosis factor a (TNFa (TNFA)), interleukin 6 (IL6), and IL1b (IL1B) and the acute phase proteins (APPs) C-reactive protein (CRP), haptoglobin, orosomucoid, YKL40 (CHI3L1), and fibrinogen were measured. Results: During GH treatment, IGF1 (median 131 (Inter-quartile range (IQR) 112–166) vs 390 (322– 524) mg/l, PZ0.002) increased together with TNFa (0.87 (0.74–1.48) vs 1.27 (0.80–1.69) ng/l, PZ0.003), IL6 (1.00 (0.83–1.55) vs 1.35 (0.80–4.28) ng/l, PZ0.045), and fibrinogen (9.2 (8.8–9.6) vs 11.1 (9.4–12.4) mM, PZ0.002). -

Anticoagulation of Children Undergoing Cardiopulmonary Bypass Is Overestimated by Current Monitoring Techniques
PAPER Anticoagulation of Children Undergoing Cardiopulmonary Bypass Is Overestimated by Current Monitoring Techniques John T. Owings, MD; Marc E. Pollock, MD; Robert C. Gosselin, MT; Kevin Ireland, RN, CCP; Jonathan S. Jahr, MD; Edward C. Larkin, MD Hypothesis: Children who undergo cardiopulmonary reserve, fibrinogen; the natural antithrombotic, anti- bypass (CPB) are proportionally more hemodiluted than thrombin; and heparin concentration. adults who undergo CPB. Current methods of monitor- ing high-dose heparin sulfate anticoagulation are depen- Results: Ten children and 10 adults completed the study. dent on fibrinogen level. Because of the decreased fi- Children had lower fibrinogen levels than adults through- brinogen levels in children, current methods of monitoring out CPB (P,.05). All adults had both therapeutic ACT and heparin anticoagulation overestimate their level of anti- Heparin Management Test levels measured throughout coagulation. CPB. Although children had therapeutic ACT levels, their Heparin Management Test levels were subtherapeutic while Design: Prospective controlled trial. undergoing CPB. The children had significantly higher thrombin-antithrombin complexes and prothrombin Main Outcome Measure: Production of thrombin (ad- fragment F1.2 than adults, indicating ongoing thrombin equacy of anticoagulation). production (P,.01). The increases in thrombin- antithrombin complexes and prothrombin fragment F1.2 Methods: Children and adults undergoing cardiac sur- in children were inversely proportional to their weight. gery who received CPB were anticoagulated in the stan- dard fashion as directed by activated clotting time Conclusions: Children undergoing CPB with heparin (ACT) results. Each subject had blood sampled at base- dosing adjusted to optimize the ACT manifest inad- line; heparinization; start of the CPB; CPB at 30, 60, equate anticoagulation (ongoing thrombin formation). -

Haptoglobin 9D91-20 30-3966/R4
HAPTOGLOBIN 9D91-20 30-3966/R4 HAPTOGLOBIN This package insert contains information to run the Haptoglobin assay on the ARCHITECT c Systems™ and the AEROSET System. NOTE: Changes Highlighted NOTE: This package insert must be read carefully prior to product use. Package insert instructions must be followed accordingly. Reliability of assay results cannot be guaranteed if there are any deviations from the instructions in this package insert. Customer Support United States: 1-877-4ABBOTT Canada: 1-800-387-8378 (English speaking customers) 1-800-465-2675 (French speaking customers) International: Call your local Abbott representative Symbols in Product Labeling Calibrators 1 through 5 Catalog number/List number Concentration Serial number Authorized Representative in the Consult instructions for use European Community Ingredients Manufacturer In vitro diagnostic medical device Temperature limitation Batch code/Lot number Use by/Expiration date Reagent 1 Reagent 2 ABBOTT LABORATORIES ABBOTT Abbott Park, IL 60064, USA Max-Planck-Ring 2 65205 Wiesbaden Germany +49-6122-580 May 2007 ©2004, 2007 Abbott Laboratories 1 NAME REAGENT HANDLING AND STORAGE HAPTOGLOBIN Reagent Handling Remove air bubbles, if present in the reagent cartridge, with a new INTENDED USE applicator stick. Alternatively, allow the reagent to sit at the appropriate The Haptoglobin assay is used for the quantitation of haptoglobin in storage temperature to allow the bubbles to dissipate. To minimize human serum or plasma. volume depletion, do not use a transfer pipette to remove the bubbles. CAUTION: Reagent bubbles may interfere with proper detection of SUMMARY AND EXPLANATION OF TEST reagent level in the cartridge, causing insufficient reagent aspiration Haptoglobin is a protein synthesized in the liver that binds with the which could impact results. -

BAFF Expression Is Increased in Patients
g in Geno nin m i ic M s ta & a P Eilertsen, J Data Mining in Genom Proteomics 2012, 3:1 D r f o Journal of o t e l DOI: 10.4172/2153-0602.1000113 o a m n r i c u s o J ISSN: 2153-0602 Data Mining in Genomics & Proteomics Research Article Open Access BAFF Expression is Increased in Patients with Lupus Nephritis and Associated with Antinucleosome Antibodies, C1 Inhibitor, Α-1-Acid- Glycoprotein and Endothelial Activation Markers G. Ø. Eilertsen¹*, M. Van Ghelue² and J.C. Nossent¹ ¹Bone and Joint research group, Department of Health Science, Medical School, University of Tromsø, Norway ²Medical Genetics Department, University Hospital North Norway, Tromsø, Norway Abstract Objectives: B cell activating factor (BAFF) inhibitor therapy has recently been approved for non-renal Systemic Lupus Erythematosus (SLE). While BAFF plays a role in experimental lupus nephritis (LN), its role human LN is not well studied. Methods: Case control study in 102 SLE patients, 30 with LN (+LN) and 72 without LN (-LN) and 31 healthy controls. We analysed BAFF mRNA expression in PBMCs (BAFF-RQ) and serum BAFF (s-BAFF) levels and investigated their relation with clinical, histological- and additional acute phase proteins. Results: s-BAFF and BAFF-RQ were increased in +LN patients compared to controls, but their expression did not correlate with ISN/RPS class, Activity- or Chronicity index on biopsy. s-BAFF correlated with levels of anti-nucleosome antibodies, C1 inhibitor and α-1-acid-glycoprotein (AGP), while BAFF-RQ correlated inversely with Factor VIII. -

Urinary Proteomics for the Early Diagnosis of Diabetic Nephropathy in Taiwanese Patients Authors
Urinary Proteomics for the Early Diagnosis of Diabetic Nephropathy in Taiwanese Patients Authors: Wen-Ling Liao1,2, Chiz-Tzung Chang3,4, Ching-Chu Chen5,6, Wen-Jane Lee7,8, Shih-Yi Lin3,4, Hsin-Yi Liao9, Chia-Ming Wu10, Ya-Wen Chang10, Chao-Jung Chen1,9,+,*, Fuu-Jen Tsai6,10,11,+,* 1 Graduate Institute of Integrated Medicine, China Medical University, Taichung, 404, Taiwan 2 Center for Personalized Medicine, China Medical University Hospital, Taichung, 404, Taiwan 3 Division of Nephrology and Kidney Institute, Department of Internal Medicine, China Medical University Hospital, Taichung, 404, Taiwan 4 Institute of Clinical Medical Science, China Medical University College of Medicine, Taichung, 404, Taiwan 5 Division of Endocrinology and Metabolism, Department of Medicine, China Medical University Hospital, Taichung, 404, Taiwan 6 School of Chinese Medicine, China Medical University, Taichung, 404, Taiwan 7 Department of Medical Research, Taichung Veterans General Hospital, Taichung, 404, Taiwan 8 Department of Social Work, Tunghai University, Taichung, 404, Taiwan 9 Proteomics Core Laboratory, Department of Medical Research, China Medical University Hospital, Taichung, 404, Taiwan 10 Human Genetic Center, Department of Medical Research, China Medical University Hospital, China Medical University, Taichung, 404, Taiwan 11 Department of Health and Nutrition Biotechnology, Asia University, Taichung, 404, Taiwan + Fuu-Jen Tsai and Chao-Jung Chen contributed equally to this work. Correspondence: Fuu-Jen Tsai, MD, PhD and Chao-Jung Chen, PhD FJ Tsai: Genetic Center, China Medical University Hospital, No.2 Yuh-Der Road, 404 Taichung, Taiwan; Telephone: 886-4-22062121 Ext. 2041; Fax: 886-4-22033295; E-mail: [email protected] CJ Chen: Graduate Institute of Integrated Medicine, China Medical University, No.91, Hsueh-Shih Road, 404, Taichung, Taiwan; Telephone: 886-4-22053366 Ext. -

Expression of Haptoglobin-Related Protein and Its Potential Role As a Tumor Antigen (Neoplasia/Breast Cancer/Acute-Phase Proteins) FRANCIS P
Proc. Natl. Acad. Sci. USA Vol. 86, pp. 1188-1192, February 1989 Biochemistry Expression of haptoglobin-related protein and its potential role as a tumor antigen (neoplasia/breast cancer/acute-phase proteins) FRANCIS P. KUHAJDA*, ASOKA I. KATUMULUWA, AND GARY R. PASTERNACK Department of Pathology, The Johns Hopkins University School of Medicine, 600 North Wolfe Street, Baltimore, MD 21205 Communicated by M. Daniel Lane, November 14, 1988 (receivedfor review August 3, 1988) ABSTRACT These studies describe the detection of a pensity for tumor invasion and early metastasis. Clinically, haptoglobin species, its characterization as the HPR gene these properties manifested as a dramatically worsened product, and its association with both pregnancy and neopla- prognosis (7-9). In Western blots (immunoblots) of fractions sia. Previous work showed that the early recurrence of human of sera from pregnant women, this antiserum detected a set breast cancer correlated with immunohistochemical staining of proteins whose properties were inconsistent with those of with a commercial antiserum ostensibly directed against preg- PAPP-A, a homotetramer composed of 200-kDa polypep- nancy-associated plasma protein A (PAPP-A). Use of this tides. The studies below describe the purification of the antiserum to guide purification ofthe putative antigen led to the unexpectedly immunoreactive protein species from the present identification and purification of a strongly immuno- plasma ofpregnant women, its identification as the HPR gene reactive protein species distinct from PAPP-A that was present product, and its relationship to the clinically important in the plasma of pregnant women at term. Unlike PAPP-A, a antigen expressed in human breast carcinoma. -

Elevated Alpha-1 Antitrypsin Is a Major Component of Glyca-Associated Risk for Future Morbidity and Mortality
bioRxiv preprint doi: https://doi.org/10.1101/309138; this version posted April 26, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Elevated alpha-1 antitrypsin is a major component of GlycA-associated risk for future morbidity and mortality Scott C. Ritchie1,2,3,4*, Johannes Kettunen5,6,7, Marta Brozynska1,2,3,4, Artika P. Nath1,8, Aki S. Havulinna6,9, Satu Männisto6, Markus Perola6,9, Veikko Salomaa6, Mika Ala-Korpela5,7,10,11,12,13, Gad Abraham1,2,3,4, Peter Würtz14,15, Michael Inouye1,2,3,4* 1Systems Genomics Lab, Baker Heart & Diabetes Institute, Melbourne, Victoria, Australia 2Department of Public Health and Primary Care, University of Cambridge, Cambridge CB1 8RN, United Kingdom 3Department of Clinical Pathology, The University of Melbourne, Parkville, VIC 3010, Australia 4School of BioSciences, The University of Melbourne, Parkville, VIC 3010, Australia 5Computational Medicine, Faculty of Medicine, University of Oulu and Biocenter Oulu, Oulu 90014, Finland 6National Institute for Health and Welfare, Helsinki 00271, Finland 7NMR Metabolomics Laboratory, School of Pharmacy, University of Eastern Finland, Kuopio 70211, Finland 8Department of Microbiology and Immunology, The University of Melbourne, Parkville, VIC 3010, Australia 9Institute for Molecular Medicine Finland, University of Helsinki, Helsinki 00014, Finland 10Population Health Science,