Non-Inferiority Clinical Trials to Establish Effectiveness
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Descriptive Statistics (Part 2): Interpreting Study Results
Statistical Notes II Descriptive statistics (Part 2): Interpreting study results A Cook and A Sheikh he previous paper in this series looked at ‘baseline’. Investigations of treatment effects can be descriptive statistics, showing how to use and made in similar fashion by comparisons of disease T interpret fundamental measures such as the probability in treated and untreated patients. mean and standard deviation. Here we continue with descriptive statistics, looking at measures more The relative risk (RR), also sometimes known as specific to medical research. We start by defining the risk ratio, compares the risk of exposed and risk and odds, the two basic measures of disease unexposed subjects, while the odds ratio (OR) probability. Then we show how the effect of a disease compares odds. A relative risk or odds ratio greater risk factor, or a treatment, can be measured using the than one indicates an exposure to be harmful, while relative risk or the odds ratio. Finally we discuss the a value less than one indicates a protective effect. ‘number needed to treat’, a measure derived from the RR = 1.2 means exposed people are 20% more likely relative risk, which has gained popularity because of to be diseased, RR = 1.4 means 40% more likely. its clinical usefulness. Examples from the literature OR = 1.2 means that the odds of disease is 20% higher are used to illustrate important concepts. in exposed people. RISK AND ODDS Among workers at factory two (‘exposed’ workers) The probability of an individual becoming diseased the risk is 13 / 116 = 0.11, compared to an ‘unexposed’ is the risk. -

Challenging Issues in Clinical Trial Design: Part 4 of a 4-Part Series on Statistics for Clinical Trials
Challenging Issues in Clinical Trial Design: Part 4 of a 4-part Series on Statistics for Clinical Trials Brief title: Challenges in Trial Design Stuart J. Pocock, PHD,* Tim C. Clayton, MSC,* Gregg W. Stone, MD† From the: *London School of Hygiene and Tropical Medicine, London, United Kingdom; †Columbia University Medical Center, New York-Presbyterian Hospital and the Cardiovascular Research Foundation, New York, New York <COR> Reprint requests and correspondence: Prof. Stuart J. Pocock, Department of Medical Statistics, London School of Hygiene and Tropical Medicine, Keppel Street, London, WC1E 7HT, United Kingdom Telephone: +44 20 7927 2413 Fax: +44 20 7637 2853 E-mail: [email protected] Disclosures: The authors declare no conflicts of interest for this paper. 1 Abstract As a sequel to last week’s article on the fundamentals of clinical trial design, this article tackles related controversial issues: noninferiority trials; the value of factorial designs; the importance and challenges of strategy trials; Data Monitoring Committees (including when to stop a trial early); and the role of adaptive designs. All topics are illustrated by relevant examples from cardiology trials. <KW>Key words: Noninferiority trials; Factorial designs; Strategy trials, Data Monitoring Committees; Statistical stopping guidelines; Adaptive designs; Randomized Controlled Trials As Topic; Abbreviations ACS = acute coronary syndrome CABG = coronary artery bypass graft CI = confidence interval CV = cardiovascular DMC = Data Monitoring Committee FDA = Food and Drug Administration MACE = major adverse cardiovascular event OMT = optimal medical therapy PCI = percutaneous coronary intervention 2 Introduction Randomized controlled trials are the cornerstone of clinical guidelines informing best therapeutic practices, however their design and interpretation may be complex and nuanced. -

Meta-Analysis of Proportions
NCSS Statistical Software NCSS.com Chapter 456 Meta-Analysis of Proportions Introduction This module performs a meta-analysis of a set of two-group, binary-event studies. These studies have a treatment group (arm) and a control group. The results of each study may be summarized as counts in a 2-by-2 table. The program provides a complete set of numeric reports and plots to allow the investigation and presentation of the studies. The plots include the forest plot, radial plot, and L’Abbe plot. Both fixed-, and random-, effects models are available for analysis. Meta-Analysis refers to methods for the systematic review of a set of individual studies with the aim to combine their results. Meta-analysis has become popular for a number of reasons: 1. The adoption of evidence-based medicine which requires that all reliable information is considered. 2. The desire to avoid narrative reviews which are often misleading. 3. The desire to interpret the large number of studies that may have been conducted about a specific treatment. 4. The desire to increase the statistical power of the results be combining many small-size studies. The goals of meta-analysis may be summarized as follows. A meta-analysis seeks to systematically review all pertinent evidence, provide quantitative summaries, integrate results across studies, and provide an overall interpretation of these studies. We have found many books and articles on meta-analysis. In this chapter, we briefly summarize the information in Sutton et al (2000) and Thompson (1998). Refer to those sources for more details about how to conduct a meta- analysis. -

EBM Notebook Numbers Needed to Treat Derived from Meta-Analysis: a Word of Caution
Evid Based Med: first published as 10.1136/ebm.8.2.36 on 1 March 2003. Downloaded from EBM notebook Numbers needed to treat derived from meta-analysis: a word of caution In clinical trials, treatment effects from binary outcomes, such the NNT will be applied, and information on the time horizon as “alive” or “dead”, can be presented in various ways (eg, rela- of the study (eg, the median follow up time). tive risk reduction [RRR] and absolute risk reduction We illustrate these points using the meta-analysis of [ARR]).1–2 (See glossary for definitions and calculations). Alter- randomised controlled trials investigating the effect of n-3 poly- natively, the number needed to treat (NNT) is an expression of the unsaturated fatty acids on clinical endpoints in patients with number of patients who need to be treated to prevent one coronary heart disease as an example.11–12 The meta-analysis additional adverse event.2–4 Mathematically, the NNT equals the found a 19% (CI 10% to 27%) RRR of overall mortality favour- reciprocal of the ARR. Many journals now report results from ing the use of n-3 polyunsaturated fatty acids. In the clinical trials using the NNT, along with 95% confidence inter- Evidence-Based Medicine structured summary of this meta- vals (CIs).5 analysis, the NNT derived from a pooled risk difference, and 11 Since its introduction,3 a debate has ensued whether based on an average baseline risk of 9.5%, is 73 (CI 49 to 147). reporting NNTs from meta-analyses is misleading.467 ACP However, this estimate does not reflect the high variability of Journal Club and Evidence-Based Medicine have devoted attention baseline risk of the included trials. -

Survival Analysis: Part I — Analysis Korean Journal of Anesthesiology of Time-To-Event
Statistical Round pISSN 2005-6419 • eISSN 2005-7563 KJA Survival analysis: Part I — analysis Korean Journal of Anesthesiology of time-to-event Junyong In1 and Dong Kyu Lee2 Department of Anesthesiology and Pain Medicine, 1Dongguk University Ilsan Hospital, Goyang, 2Guro Hospital, Korea University School of Medicine, Seoul, Korea Length of time is a variable often encountered during data analysis. Survival analysis provides simple, intuitive results concerning time-to-event for events of interest, which are not confined to death. This review introduces methods of ana- lyzing time-to-event. The Kaplan-Meier survival analysis, log-rank test, and Cox proportional hazards regression model- ing method are described with examples of hypothetical data. Keywords: Censored data; Cox regression; Hazard ratio; Kaplan-Meier method; Log-rank test; Medical statistics; Power analysis; Proportional hazards; Sample size; Survival analysis. Introduction mation of the time [2]. The Korean Journal of Anesthesiology has thus far published In a clinical trial or clinical study, an investigational treat- several papers using survival analysis for clinical outcomes: a ment is administered to subjects, and the resulting outcome data comparison of 90-day survival rate for pressure ulcer develop- are collected and analyzed after a certain period of time. Most ment or non-development after surgery under general anesthesia statistical analysis methods do not include the length of time [3], a comparison of postoperative 5-year recurrence rate in as a variable, and analysis is made only on the trial outcomes patients with breast cancer depending on the type of anesthetic upon completion of the study period, as specified in the study agent used [4], and a comparison of airway intubation success protocol. -
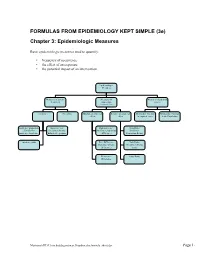
FORMULAS from EPIDEMIOLOGY KEPT SIMPLE (3E) Chapter 3: Epidemiologic Measures
FORMULAS FROM EPIDEMIOLOGY KEPT SIMPLE (3e) Chapter 3: Epidemiologic Measures Basic epidemiologic measures used to quantify: • frequency of occurrence • the effect of an exposure • the potential impact of an intervention. Epidemiologic Measures Measures of disease Measures of Measures of potential frequency association impact (“Measures of Effect”) Incidence Prevalence Absolute measures of Relative measures of Attributable Fraction Attributable Fraction effect effect in exposed cases in the Population Incidence proportion Incidence rate Risk difference Risk Ratio (Cumulative (incidence density, (Incidence proportion (Incidence Incidence, Incidence hazard rate, person- difference) Proportion Ratio) Risk) time rate) Incidence odds Rate Difference Rate Ratio (Incidence density (Incidence density difference) ratio) Prevalence Odds Ratio Difference Macintosh HD:Users:buddygerstman:Dropbox:eks:formula_sheet.doc Page 1 of 7 3.1 Measures of Disease Frequency No. of onsets Incidence Proportion = No. at risk at beginning of follow-up • Also called risk, average risk, and cumulative incidence. • Can be measured in cohorts (closed populations) only. • Requires follow-up of individuals. No. of onsets Incidence Rate = ∑person-time • Also called incidence density and average hazard. • When disease is rare (incidence proportion < 5%), incidence rate ≈ incidence proportion. • In cohorts (closed populations), it is best to sum individual person-time longitudinally. It can also be estimated as Σperson-time ≈ (average population size) × (duration of follow-up). Actuarial adjustments may be needed when the disease outcome is not rare. • In an open populations, Σperson-time ≈ (average population size) × (duration of follow-up). Examples of incidence rates in open populations include: births Crude birth rate (per m) = × m mid-year population size deaths Crude mortality rate (per m) = × m mid-year population size deaths < 1 year of age Infant mortality rate (per m) = × m live births No. -

CHAPTER 3 Comparing Disease Frequencies
© Smartboy10/DigitalVision Vectors/Getty Images CHAPTER 3 Comparing Disease Frequencies LEARNING OBJECTIVES By the end of this chapter the reader will be able to: ■ Organize disease frequency data into a two-by-two table. ■ Describe and calculate absolute and relative measures of comparison, including rate/risk difference, population rate/risk difference, attributable proportion among the exposed and the total population and rate/risk ratio. ■ Verbally interpret each absolute and relative measure of comparison. ■ Describe the purpose of standardization and calculate directly standardized rates. ▸ Introduction Measures of disease frequency are the building blocks epidemiologists use to assess the effects of a disease on a population. Comparing measures of disease frequency organizes these building blocks in a meaningful way that allows one to describe the relationship between a characteristic and a disease and to assess the public health effect of the exposure. Disease frequencies can be compared between different populations or between subgroups within a population. For example, one might be interested in comparing disease frequencies between residents of France and the United States or between subgroups within the U.S. population according to demographic characteristics, such as race, gender, or socio- economic status; personal habits, such as alcoholic beverage consump- tion or cigarette smoking; and environmental factors, such as the level of air pollution. For example, one might compare incidence rates of coronary heart disease between residents of France and those of the United States or 57 58 Chapter 3 Comparing Disease Frequencies among U.S. men and women, Blacks and Whites, alcohol drinkers and nondrinkers, and areas with high and low pollution levels. -

Outcome Reporting Bias in COVID-19 Mrna Vaccine Clinical Trials
medicina Perspective Outcome Reporting Bias in COVID-19 mRNA Vaccine Clinical Trials Ronald B. Brown School of Public Health and Health Systems, University of Waterloo, Waterloo, ON N2L3G1, Canada; [email protected] Abstract: Relative risk reduction and absolute risk reduction measures in the evaluation of clinical trial data are poorly understood by health professionals and the public. The absence of reported absolute risk reduction in COVID-19 vaccine clinical trials can lead to outcome reporting bias that affects the interpretation of vaccine efficacy. The present article uses clinical epidemiologic tools to critically appraise reports of efficacy in Pfzier/BioNTech and Moderna COVID-19 mRNA vaccine clinical trials. Based on data reported by the manufacturer for Pfzier/BioNTech vaccine BNT162b2, this critical appraisal shows: relative risk reduction, 95.1%; 95% CI, 90.0% to 97.6%; p = 0.016; absolute risk reduction, 0.7%; 95% CI, 0.59% to 0.83%; p < 0.000. For the Moderna vaccine mRNA-1273, the appraisal shows: relative risk reduction, 94.1%; 95% CI, 89.1% to 96.8%; p = 0.004; absolute risk reduction, 1.1%; 95% CI, 0.97% to 1.32%; p < 0.000. Unreported absolute risk reduction measures of 0.7% and 1.1% for the Pfzier/BioNTech and Moderna vaccines, respectively, are very much lower than the reported relative risk reduction measures. Reporting absolute risk reduction measures is essential to prevent outcome reporting bias in evaluation of COVID-19 vaccine efficacy. Keywords: mRNA vaccine; COVID-19 vaccine; vaccine efficacy; relative risk reduction; absolute risk reduction; number needed to vaccinate; outcome reporting bias; clinical epidemiology; critical appraisal; evidence-based medicine Citation: Brown, R.B. -

Delineating Virulence of Vibrio Campbellii
www.nature.com/scientificreports OPEN Delineating virulence of Vibrio campbellii: a predominant luminescent bacterial pathogen in Indian shrimp hatcheries Sujeet Kumar1*, Chandra Bhushan Kumar1,2, Vidya Rajendran1, Nishawlini Abishaw1, P. S. Shyne Anand1, S. Kannapan1, Viswas K. Nagaleekar3, K. K. Vijayan1 & S. V. Alavandi1 Luminescent vibriosis is a major bacterial disease in shrimp hatcheries and causes up to 100% mortality in larval stages of penaeid shrimps. We investigated the virulence factors and genetic identity of 29 luminescent Vibrio isolates from Indian shrimp hatcheries and farms, which were earlier presumed as Vibrio harveyi. Haemolysin gene-based species-specifc multiplex PCR and phylogenetic analysis of rpoD and toxR identifed all the isolates as V. campbellii. The gene-specifc PCR revealed the presence of virulence markers involved in quorum sensing (luxM, luxS, cqsA), motility (faA, lafA), toxin (hly, chiA, serine protease, metalloprotease), and virulence regulators (toxR, luxR) in all the isolates. The deduced amino acid sequence analysis of virulence regulator ToxR suggested four variants, namely A123Q150 (AQ; 18.9%), P123Q150 (PQ; 54.1%), A123P150 (AP; 21.6%), and P123P150 (PP; 5.4% isolates) based on amino acid at 123rd (proline or alanine) and 150th (glutamine or proline) positions. A signifcantly higher level of the quorum-sensing signal, autoinducer-2 (AI-2, p = 2.2e−12), and signifcantly reduced protease activity (p = 1.6e−07) were recorded in AP variant, whereas an inverse trend was noticed in the Q150 variants AQ and PQ. The pathogenicity study in Penaeus (Litopenaeus) vannamei juveniles revealed that all the isolates of AQ were highly pathogenic with Cox proportional hazard ratio 15.1 to 32.4 compared to P150 variants; PP (5.4 to 6.3) or AP (7.3 to 14). -

Should the Randomistas (Continue To) Rule?
Should the Randomistas (Continue to) Rule? Martin Ravallion Abstract The rising popularity of randomized controlled trials (RCTs) in development applications has come with continuing debates on the pros and cons of this approach. The paper revisits the issues. While RCTs have a place in the toolkit for impact evaluation, an unconditional preference for RCTs as the “gold standard” is questionable. The statistical case is unclear on a priori grounds; a stronger ethical defense is often called for; and there is a risk of distorting the evidence-base for informing policymaking. Going forward, pressing knowledge gaps should drive the questions asked and how they are answered, not the methodological preferences of some researchers. The gold standard is the best method for the question at hand. Keywords: Randomized controlled trials, bias, ethics, external validity, ethics, development policy JEL: B23, H43, O22 Working Paper 492 August 2018 www.cgdev.org Should the Randomistas (Continue to) Rule? Martin Ravallion Department of Economics, Georgetown University François Roubaud encouraged the author to write this paper. For comments the author is grateful to Sarah Baird, Mary Ann Bronson, Caitlin Brown, Kevin Donovan, Markus Goldstein, Miguel Hernan, Emmanuel Jimenez, Madhulika Khanna, Nishtha Kochhar, Andrew Leigh, David McKenzie, Berk Özler, Dina Pomeranz, Lant Pritchett, Milan Thomas, Vinod Thomas, Eva Vivalt, Dominique van de Walle and Andrew Zeitlin. Staff of the International Initiative for Impact Evaluation kindly provided an update to their database on published impact evaluations and helped with the author’s questions. Martin Ravallion, 2018. “Should the Randomistas (Continue to) Rule?.” CGD Working Paper 492. Washington, DC: Center for Global Development. -

Clinical Epidemiologic Definations
CLINICAL EPIDEMIOLOGIC DEFINATIONS 1. STUDY DESIGN Case-series: Report of a number of cases of disease. Cross-sectional study: Study design, concurrently measure outcome (disease) and risk factor. Compare proportion of diseased group with risk factor, with proportion of non-diseased group with risk factor. Case-control study: Retrospective comparison of exposures of persons with disease (cases) with those of persons without the disease (controls) (see Retrospective study). Retrospective study: Study design in which cases where individuals who had an outcome event in question are collected and analyzed after the outcomes have occurred (see also Case-control study). Cohort study: Follow-up of exposed and non-exposed defined groups, with a comparison of disease rates during the time covered. Prospective study: Study design where one or more groups (cohorts) of individuals who have not yet had the outcome event in question are monitored for the number of such events, which occur over time. Randomized controlled trial: Study design where treatments, interventions, or enrollment into different study groups are assigned by random allocation rather than by conscious decisions of clinicians or patients. If the sample size is large enough, this study design avoids problems of bias and confounding variables by assuring that both known and unknown determinants of outcome are evenly distributed between treatment and control groups. Bias (Syn: systematic error): Deviation of results or inferences from the truth, or processes leading to such deviation. See also Referral Bias, Selection Bias. Recall bias: Systematic error due to the differences in accuracy or completeness of recall to memory of past events or experiences. -

Enrichment Strategies for Clinical Trials to Support Determination of Effectiveness of Human Drugs and Biological Products Guidance for Industry
Enrichment Strategies for Clinical Trials to Support Determination of Effectiveness of Human Drugs and Biological Products Guidance for Industry U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) March 2019 Clinical/Medical Enrichment Strategies for Clinical Trials to Support Determination of Effectiveness of Human Drugs and Biological Products Guidance for Industry Additional copies are available from: Office of Communications, Division of Drug Information Center for Drug Evaluation and Research Food and Drug Administration 10001 New Hampshire Ave., Hillandale Bldg., 4th Floor Silver Spring, MD 20993-0002 Phone: 855-543-3784 or 301-796-3400; Fax: 301-431-6353; Email: [email protected] https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm and/or Office of Communication, Outreach, and Development Center for Biologics Evaluation and Research Food and Drug Administration 10903 New Hampshire Ave., Bldg. 71, rm. 3128 Silver Spring, MD 20993-0002 Phone: 800-835-4709 or 240-402-8010; Email: [email protected] https://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) March 2019 Clinical/Medical TABLE OF CONTENTS I. INTRODUCTION............................................................................................................