Variability in Dentofacial Phenotypes in Four Families with WNT10A Mutations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A High-Fat/High-Sucrose Diet Induces WNT4 Expression in Mouse Pancreatic Β-Cells
This is “Advance Publication Article” Kurume Medical Journal, 65, 00-00, 2018 Original Article A High-Fat/High-Sucrose Diet Induces WNT4 Expression in Mouse Pancreatic β-cells YAYOI KURITA, TSUYOSHI OHKI, ERI SOEJIMA, XIAOHONG YUAN, SATOMI KAKINO, NOBUHIKO WADA, TOSHIHIKO HASHINAGA, HITOMI NAKAYAMA, JUNICHI TANI, YUJI TAJIRI, YUJI HIROMATSU, KENTARO YAMADA* AND MASATOSHI NOMURA Division of Endocrinology and Metabolism, Department of Internal Medicine, Kurume University School of Medicine, Kurume 830-0011, Japan, *Diabetes Center of Asakura Medical Association Hospital, Asakura 838-0069, Japan Received 12 February 2018, accepted 1 May 2018 J-STAGE advance publication 11 March 2019 Edited by MAKOTO TAKANO Summary: Aims/Introduction: Several lines of evidence suggest that dysregulation of the WNT signaling pathway is involved in the pathogenesis of type 2 diabetes. This study was performed to elucidate the effects of a high-fat/high-sucrose (HF/HS) diet on pancreatic islet functions in relation to modulation of WNT ligand expression in β-cells. Materials and Methods: Mice were fed either standard mouse chow or a HF/HS diet from 8 weeks of age. At 20 weeks of age, intraperitoneal glucose tolerance tests were performed in both groups of mice, followed by euthanasia and isolation of pancreatic islets. WNT-related gene expression in islets and MIN6 cells was measured by quantitative real-time RT-PCR. To explore the direct effects of WNT signals on pancreatic β-cells, MIN6 cells were exposed to recombinant mouse WNT4 protein (rmWNT4) for 48 h, and glucose-induced insulin secre- tion was measured. Furthermore, Wnt4 siRNAs were transfected into MIN6 cells, and cell viability and insulin secretion were measured in control and Wnt4 siRNA-transfected MIN6 cells. -

WNT10A Gene Wnt Family Member 10A
WNT10A gene Wnt family member 10A Normal Function The WNT10A gene is part of a large family of WNT genes, which play critical roles in development starting before birth. These genes provide instructions for making proteins that participate in chemical signaling pathways in the body. Wnt signaling controls the activity of certain genes and regulates the interactions between cells during embryonic development. The protein produced from the WNT10A gene plays a role in the development of many parts of the body. It appears to be essential for the formation of tissues that arise from an embryonic cell layer called the ectoderm. These tissues include the skin, hair, nails, teeth, and sweat glands. Researchers believe that the WNT10A protein is particularly important for the formation and shaping of both baby (primary) teeth and adult ( permanent) teeth. Health Conditions Related to Genetic Changes Hypohidrotic ectodermal dysplasia Several mutations in the WNT10A gene have been found to cause hypohidrotic ectodermal dysplasia, the most common form of ectodermal dysplasia. Starting before birth, ectodermal dysplasias result in the abnormal development of the skin, hair, nails, teeth, and sweat glands. Hypohidrotic ectodermal dysplasia is characterized by a reduced ability to sweat (hypohidrosis), sparse scalp and body hair (hypotrichosis), and several missing teeth (hypodontia) or teeth that are malformed. WNT10A gene mutations account for about 5 percent of all cases of hypohidrotic ectodermal dysplasia. Most of the WNT10A gene mutations associated with hypohidrotic ectodermal dysplasia change single protein building blocks (amino acids) in the WNT10A protein, which impairs its function. The resulting shortage of functional WNT10A protein disrupts Wnt signaling during the development of ectodermal tissues, particularly the teeth. -

Theranostics WNT6 Is a Novel Oncogenic Prognostic Biomarker In
Theranostics 2018, Vol. 8, Issue 17 4805 Ivyspring International Publisher Theranostics 2018; 8(17): 4805-4823. doi: 10.7150/thno.25025 Research Paper WNT6 is a novel oncogenic prognostic biomarker in human glioblastoma Céline S. Gonçalves1,2, Joana Vieira de Castro1,2, Marta Pojo1,2, Eduarda P. Martins1,2, Sandro Queirós1,2, Emmanuel Chautard3,4, Ricardo Taipa5, Manuel Melo Pires5, Afonso A. Pinto6, Fernando Pardal7, Carlos Custódia8, Cláudia C. Faria8,9, Carlos Clara10, Rui M. Reis1,2,10, Nuno Sousa1,2, Bruno M. Costa1,2 1. Life and Health Sciences Research Institute (ICVS), School of Medicine, University of Minho, Campus Gualtar, 4710-057 Braga, Portugal 2. ICVS/3B’s - PT Government Associate Laboratory, Braga/Guimarães, Portugal 3. Université Clermont Auvergne, INSERM, U1240 IMoST, 63000 Clermont Ferrand, France 4. Pathology Department, Université Clermont Auvergne, Centre Jean Perrin, 63011 Clermont-Ferrand, France 5. Neuropathology Unit, Department of Neurosciences, Centro Hospitalar do Porto, Porto, Portugal 6. Department of Neurosurgery, Hospital Escala Braga, Sete Fontes - São Victor 4710-243 Braga, Portugal 7. Department of Pathology, Hospital Escala Braga, Sete Fontes - São Victor 4710-243 Braga, Portugal 8. Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Lisbon, Portugal 9. Neurosurgery Department, Hospital de Santa Maria, Centro Hospitalar Lisboa Norte (CHLN), Lisbon, Portugal 10. Molecular Oncology Research Center, Barretos Cancer Hospital, Barretos - S. Paulo, Brazil. Corresponding author: Bruno M. Costa, Life and Health Sciences Research Institute (ICVS), School of Medicine, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal. Email: [email protected]; Phone: (+351)253604837; Fax: (+351)253604831 © Ivyspring International Publisher. -

Wnt-Independent and Wnt-Dependent Effects of APC Loss on the Chemotherapeutic Response
International Journal of Molecular Sciences Review Wnt-Independent and Wnt-Dependent Effects of APC Loss on the Chemotherapeutic Response Casey D. Stefanski 1,2 and Jenifer R. Prosperi 1,2,3,* 1 Department of Biological Sciences, University of Notre Dame, Notre Dame, IN 46617, USA; [email protected] 2 Mike and Josie Harper Cancer Research Institute, South Bend, IN 46617, USA 3 Department of Biochemistry and Molecular Biology, Indiana University School of Medicine-South Bend, South Bend, IN 46617, USA * Correspondence: [email protected]; Tel.: +1-574-631-4002 Received: 30 September 2020; Accepted: 20 October 2020; Published: 22 October 2020 Abstract: Resistance to chemotherapy occurs through mechanisms within the epithelial tumor cells or through interactions with components of the tumor microenvironment (TME). Chemoresistance and the development of recurrent tumors are two of the leading factors of cancer-related deaths. The Adenomatous Polyposis Coli (APC) tumor suppressor is lost in many different cancers, including colorectal, breast, and prostate cancer, and its loss correlates with a decreased overall survival in cancer patients. While APC is commonly known for its role as a negative regulator of the WNT pathway, APC has numerous binding partners and functional roles. Through APC’s interactions with DNA repair proteins, DNA replication proteins, tubulin, and other components, recent evidence has shown that APC regulates the chemotherapy response in cancer cells. In this review article, we provide an overview of some of the cellular processes in which APC participates and how they impact chemoresistance through both epithelial- and TME-derived mechanisms. Keywords: adenomatous polyposis coli; chemoresistance; WNT signaling 1. -

Deregulated Wnt/Β-Catenin Program in High-Risk Neuroblastomas Without
Oncogene (2008) 27, 1478–1488 & 2008 Nature Publishing Group All rights reserved 0950-9232/08 $30.00 www.nature.com/onc ONCOGENOMICS Deregulated Wnt/b-catenin program in high-risk neuroblastomas without MYCN amplification X Liu1, P Mazanek1, V Dam1, Q Wang1, H Zhao2, R Guo2, J Jagannathan1, A Cnaan2, JM Maris1,3 and MD Hogarty1,3 1Division of Oncology, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA; 2Department of Biostatistics and Epidemiology, University of Pennsylvania School of Medicine, Philadelphia, PA, USA and 3Department of Pediatrics, University of Pennsylvania School of Medicine, Philadelphia, PA, USA Neuroblastoma (NB) is a frequently lethal tumor of Introduction childhood. MYCN amplification accounts for the aggres- sive phenotype in a subset while the majority have no Neuroblastoma (NB) is a childhood embryonal malig- consistently identified molecular aberration but frequently nancy arising in the peripheral sympathetic nervous express MYC at high levels. We hypothesized that acti- system. Half of all children with NB present with features vated Wnt/b-catenin (CTNNB1) signaling might account that define their tumorsashigh riskwith poor overall for this as MYC is a b-catenin transcriptional target and survival despite intensive therapy (Matthay et al., 1999). multiple embryonal and neural crest malignancies have A subset of these tumors are characterized by high-level oncogenic alterations in this pathway. NB cell lines without genomic amplification of the MYCN proto-oncogene MYCN amplification express higher levels of MYC and (Matthay et al., 1999) but the remainder have no b-catenin (with aberrant nuclear localization) than MYCN- consistently identified aberration to account for their amplified cell lines. -

Towards an Integrated View of Wnt Signaling in Development Renée Van Amerongen and Roel Nusse*
HYPOTHESIS 3205 Development 136, 3205-3214 (2009) doi:10.1242/dev.033910 Towards an integrated view of Wnt signaling in development Renée van Amerongen and Roel Nusse* Wnt signaling is crucial for embryonic development in all animal Notably, components at virtually every level of the Wnt signal species studied to date. The interaction between Wnt proteins transduction cascade have been shown to affect both β-catenin- and cell surface receptors can result in a variety of intracellular dependent and -independent responses, depending on the cellular responses. A key remaining question is how these specific context. As we discuss below, this holds true for the Wnt proteins responses take shape in the context of a complex, multicellular themselves, as well as for their receptors and some intracellular organism. Recent studies suggest that we have to revise some of messengers. Rather than concluding that these proteins are shared our most basic ideas about Wnt signal transduction. Rather than between pathways, we instead propose that it is the total net thinking about Wnt signaling in terms of distinct, linear, cellular balance of signals that ultimately determines the response of the signaling pathways, we propose a novel view that considers the receiving cell. In the context of an intact and developing integration of multiple, often simultaneous, inputs at the level organism, cells receive multiple, dynamic, often simultaneous and of both Wnt-receptor binding and the downstream, sometimes even conflicting inputs, all of which are integrated to intracellular response. elicit the appropriate cell behavior in response. As such, the different signaling pathways might thus be more intimately Introduction intertwined than previously envisioned. -

Role of DNA Methylation in Adipogenesis
Georgia State University ScholarWorks @ Georgia State University Biology Theses Department of Biology Summer 8-12-2014 Role of DNA Methylation in Adipogenesis Yii-Shyuan Chen Follow this and additional works at: https://scholarworks.gsu.edu/biology_theses Recommended Citation Chen, Yii-Shyuan, "Role of DNA Methylation in Adipogenesis." Thesis, Georgia State University, 2014. https://scholarworks.gsu.edu/biology_theses/57 This Thesis is brought to you for free and open access by the Department of Biology at ScholarWorks @ Georgia State University. It has been accepted for inclusion in Biology Theses by an authorized administrator of ScholarWorks @ Georgia State University. For more information, please contact [email protected]. ROLE OF DNA METHYLATION IN ADIPOGENESIS by YII-SHYUAN CHEN Under the Direction of Bingzhong Xue ABSTRACT The increase in the prevalence of obesity and obesity-related diseases has caused greater attention to be placed on the molecular mechanisms controlling adipogenesis. In this study, we studied the role of 5-aza-2'-deoxycytidine (5-Aza-dC), an inhibitor of DNA methylation, on adipocyte differentiation. We found that inhibiting DNA methylation by 5-Aza-dC significantly inhibited adipocyte differentiation whereas promoting osteoblastogenesis. Wnt10a was up- regulated by 5-Aza-dC treatment and it was suggested that Wnt10a might play a vital role in suppressing adipogenesis and promoting osteoblastogenesis by inhibiting DNA methylation. In 3T3-L1 cells, Wnt signaling inhibitor IWP-2 was found to reverse the inhibitory effect of 5-Aza- dC on Adipocyte differentiation, whereas in mesenchymal stem cell line, ST2 cells, IWP-2 treatment reversed the effect of 5-Aza-dC on promoting osteoblastogenesis. -

Wnt4/B2catenin Signaling in Medullary Kidney Myofibroblasts
BASIC RESEARCH www.jasn.org Wnt4/b2Catenin Signaling in Medullary Kidney Myofibroblasts † †‡ | Derek P. DiRocco,* Akio Kobayashi,* Makoto M. Taketo,§ Andrew P. McMahon, and †‡ Benjamin D. Humphreys* *Renal Division, Brigham and Women’s Hospital, Boston, Massachusetts; †Harvard Medical School, Boston, Massachusetts; ‡Harvard Stem Cell Institute, Cambridge, Massachusetts; §Department of Pharmacology, Graduate School of Medicine, Kyoto University, Yoshida-Konoé-cho, Sakyo, Kyoto, Japan; and |Department of Stem Cell Biology and Regenerative Medicine, Eli and Edythe Broad-CIRM Center for Regenerative Medicine and Stem Cell Research, Keck School of Medicine of the University of Southern California, Los Angeles, California ABSTRACT Injury to the adult kidney induces a number of developmental genes thought to regulate repair, including Wnt4. During kidney development, early nephron precursors and medullary stroma both express Wnt4, where it regulates epithelialization and controls smooth muscle fate, respectively. Expression patterns and roles for Wnt4 in the adult kidney, however, remain unclear. In this study, we used reporters, lineage analysis, and conditional knockout or activation of the Wnt/b-catenin pathway to investigate Wnt4 in the adult kidney. Proliferating, medullary, interstitial myofibroblasts strongly expressed Wnt4 during renal fibrosis, whereas tubule epithelia, except for the collecting duct, did not. Exogenous Wnt4 drove myofi- broblast differentiation of a pericyte-like cell line, suggesting that Wnt4 might regulate pericyte-to-myo- fibroblast transition through autocrine signaling. However, conditional deletion of Wnt4 in interstitial cells did not reduce myofibroblast proliferation, cell number, or myofibroblast gene expression during fibrosis. Because the injured kidney expresses multiple Wnt ligands that might compensate for the absence of Wnt4, we generated a mouse model with constitutive activation of canonical Wnt/b-catenin signaling in interstitial pericytes and fibroblasts. -

Induction of Apoptosis Increases Expression of Non-Canonical WNT Genes in Myeloid Leukemia Cell Lines
1563-1569 7/11/07 19:04 Page 1563 ONCOLOGY REPORTS 18: 1563-1569, 2007 Induction of apoptosis increases expression of non-canonical WNT genes in myeloid leukemia cell lines HAKKI OGUN SERCAN1, MELEK PEHLIVAN1, OZLENEN SIMSEK1, HALIL ATES2 and ZEYNEP SERCAN1 Depatments of 1Medical Biology and Genetics, 2Hematology/Oncology, Dokuz Eylul University Faculty of Medicine, Izmir, Turkey Received May 3, 2007; Accepted July 20, 2007 Abstract. With the aim of determining the differential and cyclin D. The presence or exclusion of co-receptors (low expression of WNT and FZD genes, before and after density lipoprotein receptor-related protein 5 and 6-LRP5/6) induction of apoptosis in BCR-ABL positive cells, we treated or secreted Wnt antagonists (soluble frizzled related proteins the myeloid cell line K562 and control cell line HL60 with - sFRPs- and Dickkopf proteins) increases the complexity of imatinib mesylate and etoposide, and analyzed relative the molecular mechanisms underlying cellular outcomes. mRNA expression levels of WNT, FZD and sFRP genes Non-canonical pathway activation is independent of ß- under normal and apoptotic conditions by real-time RT-PCR. catenin. Two non-canonical pathways have also been defined: We observed marked increase in mRNA levels of FZD4, the planar cell polarity (PCP) and the Wnt/Ca2+ pathways (3-6). FZD5, FZD7 and WNT5b, correlating with apoptotic activity There is an overlap of proposed Wnt and Fzd proteins and and independent of the agent or cell line used. Our results secondary messengers participating in these non-canonical suggest the involvement of non-canonical Wnt signaling in pathways, complicating further the interpretation of experi- executing programmed cell death in myeloid cell lines. -

Novel Missense Mutations in the AXIN2 Gene Associated with Non
a r c h i v e s o f o r a l b i o l o g y 5 9 ( 2 0 1 4 ) 3 4 9 – 3 5 3 Available online at www.sciencedirect.com ScienceDirect journal homepage: http://www.elsevier.com/locate/aob Novel missense mutations in the AXIN2 gene associated with non-syndromic oligodontia a,1 a,1 b a Singwai Wong , Haochen Liu , Baojing Bai , Huaiguang Chang , c,d e a, a, Hongshan Zhao , Yixiang Wang , Dong Han **, Hailan Feng * a Department of Prosthodontics, Peking University School and Hospital of Stomatology, Beijing, China b Department of Prosthodontics, Beijing Stomatology Hospital, Beijing, China c Department of Medical Genetics, Peking University Health Science Center, Beijing, China d Peking University Center for Human Disease Genomics, Peking University Health Science Center, Beijing, China e Central Laboratory, Peking University School and Hospital of Stomatology, Beijing, China a r t i c l e i n f o a b s t r a c t Article history: Objective: Oligodontia, which is the congenital absence of six or more permanent teeth Accepted 23 December 2013 excluding third molars, may contribute to masticatory dysfunction, speech alteration, aesthetic problems and malocclusion. To date, mutations in EDA, AXIN2, MSX1, PAX9, Keywords: WNT10A, EDAR, EDARADD, NEMO and KRT 17 are known to associate with non-syndromic oligodontia. The aim of the study was to search for AXIN2 mutations in 96 patients with non- Non-syndromic oligodontia syndromic oligodontia. Mutation screening AXIN2 Design: We performed mutation analysis of 10 exons of the AXIN2 gene in 96 patients with isolated non-syndromic oligodontia. -

The WNT10A Gene in Ectodermal Dysplasias and Selective Tooth Agenesis Gabriele Mues,1* John Bonds,1 Lilin Xiang,1 Alexandre R
RESEARCH ARTICLE The WNT10A Gene in Ectodermal Dysplasias and Selective Tooth Agenesis Gabriele Mues,1* John Bonds,1 Lilin Xiang,1 Alexandre R. Vieira,2 Figen Seymen,3 Ophir Klein,4,5,6 and Rena N. D’Souza1 1Department of Biomedical Sciences, Texas A&M University-HSC Baylor College of Dentistry, Dallas, Texas 2Department of Oral Biology, University of Pittsburgh School of Dental Medicine, Pittsburgh, Pennsylvania 3University of Istanbul Faculty of Dentistry, Istanbul, Turkey 4Department of Orofacial Sciences, University of California San Francisco, San Francisco, California 5Department of Pediatrics, University of California San Francisco, San Francisco, California 6Institute for Human Genetics and Regeneration Medicine, University of California San Francisco, San Francisco, California Manuscript Received: 8 October 2013; Manuscript Accepted: 30 January 2014 Mutations in the WNT10A gene were first detected in the rare syndrome odonto-onycho-dermal dysplasia (OODD, How to Cite this Article: OMIM257980) but have now also been found to cause about Mues G, Bonds J, Xiang L, Vieira AR, 35–50% of selective tooth agenesis (STHAG4, OMIM150400), a Seymen F, Klein O, D’Souza RN. 2014. common disorder that mostly affects the permanent dentition. The WNT10A gene in ectodermal In our random sample of tooth agenesis patients, 40% had at least dysplasias and selective tooth agenesis. one mutation in the WNT10A gene. The WNT10A Phe228Ile variant alone reached an allele frequency of 0.21 in the tooth Am J Med Genet Part A 164A:2455–2460. agenesis cohort, about 10 times higher than the allele frequency reported in large SNP databases for Caucasian populations. Patients with bi-allelic WNT10A mutations have severe tooth syndrome (OMIM 224750) which additionally features eyelid cysts agenesis while heterozygous individuals are either unaffected or and predisposition to adnexal skin tumors. -
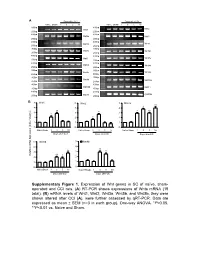
Supplementary Figure 1. Expression of Wnt Genes in SC of Naïve
A Days after CCI Days after CCI Naive Sham 1 3 5 10 Naive Sham 1 3 5 10 500bp 400bp Wnt1 Wnt2 250bp 250bp 400bp 400bp Wnt2b Wnt3 250bp 250bp 400bp 400bp Wnt3a Wnt4 250bp 250bp 300bp 400bp Wnt5a Wnt5b 200bp 250bp 400bp 400bp Wnt6 Wnt7a 250bp 250bp 400bp 400bp Wnt7b Wnt8a 250bp 250bp 400bp 350bp Wnt8b Wnt9a 250bp 200bp 400bp 400bp Wnt9b Wnt10a 250bp 250bp 400bp 400bp Wnt10b Wnt11 250bp 250bp 400bp 400bp Wnt16 GAPDH 250bp 250bp B 5 Wnt1 5 Wnt2 5 Wnt3a ** 4 4 4 * ** ** 3 3 * 3 * * 2 2 * 2 1 1 1 0 0 0 NaïveSham 1 3 5 10 Naïve Sham 1 3 5 10 Naïve Sham 1 3 5 10 Days after CCI Days after CCI Days after CCI 5 Wnt5b 5 Wnt8b 4 4 ** 3 ** 3 ** Relative mRNA Expression (fold of change) ** 2 * 2 1 1 0 0 NaïveSham 1 3 5 10 NaïveSham 1 3 5 10 Days after CCI Days after CCI Supplementary Figure 1 A B Negative Weak Moderate Strong Negative Weak Moderate Strong Large-sized cells 100% 100% 80% 80% 60% 60% 40% 20% 40% 0% 20% Sham CCI-1d CCI-14d Medium-sized cells Wnt3a immunoreactivity cells 0% 100% CGRP(+) IB4(+) 80% Small cells 60% 40% 20% 0% Sham CCI-1d CCI-14d Wnt3a immunoreactivity cells Small cells 100% 80% 60% 40% 20% 0% Sham CCI-1d CCI-14d Fig. S2 4 Fz1 4 Fz3 4 Fz4 4 Fz5 ** 3 3 ** 3 3 2 ** 2 2 ** 2 * 1 1 1 1 0 0 0 0 NaïveSham 1 5 10 NaïveSham 1 5 10 Naïve Sham 1 5 10 NaïveSham 1 5 10 Days after CCI Days after CCI Days after CCI Days after CCI 4 Fz6 4 Fz7 4 Fz8 4 Fz9 (Fold of Change) of (Fold 3 3 3 ** 3 Relative mRNA Expression Expression mRNA Relative ** 2 2 2 ** 2 * 1 1 1 1 0 0 0 0 NaïveSham 1 5 10 NaïveSham 1 5 10 Naïve Sham 1d 5d 10d NaïveSham 1