Genetic Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Case Study: Testing for Genetic Disorders
Case study: Testing for genetic disorders Purpose: A genetic disorder is a disorder that is caused by an abnormality (or several abnormalities) in a person’s genome. Genetic disorders are often hereditary, which means they are passed down to a child from its parents. The most common genetic disorder is familial hypercholesterolaemia (a genetic predisposition to high cholesterol levels), which is thought to affect 1 in 250 people in the UK. Other disorders, such as cystic fibrosis, are rarer although they can be relatively common in particular ethnic groups; for instance, the incidence of cystic fibrosis in Scotland is almost double the global average. Collectively genetic disorders affect lots of people because there are more than 10,000 different types worldwide. Genetic testing is used to inform people whether they have a disorder, or if there is a chance they might pass a disorder on to their children. Photo credit: jxfzsy/ iStockphoto Photo credit: monkeybusiness/ iStockphoto Approaches to identifying genetic disorders In the UK, if a person is concerned they might have, or might develop, a genetic disorder based on their family history, they can: Go to their GP, who might refer them for genetic testing. Patients usually provide a DNA sample, which is then analysed for a specific abnormality. Usually a single gene will be tested, but whole-genome sequencing approaches are being developed. The 100,000 Genomes Project, for example, aims to study whole-genome sequences from patients with cancer and rare disease Use a commercial genetic testing service. In this case, customers collect samples of their own DNA and send them away for analysis and interpretation Take no action. -

DOI: 10.4274/Jcrpe.Galenos.2021.2020.0175
DOI: 10.4274/jcrpe.galenos.2021.2020.0175 Case report A novel SCNN1A variation in a patient with autosomal-recessive pseudohypoaldosteronism type 1 Mohammed Ayed Huneif1*, Ziyad Hamad AlHazmy2, Anas M. Shoomi 3, Mohammed A. AlGhofely 3, Dr Humariya Heena 5, Aziza M. Mushiba 4, Abdulhamid AlSaheel3 1Pediatric Endocrinologist at Najran university hospital, Najran Saudi Arabia. 2 Pediatric Endocrinologist at Al yamammah hospital, , Riyadh, Saudi Arabia. 3 Pediatric Endocrinologist at Pediatric endocrine department,. Obesity, Endocrine, and Metabolism Center, , King Fahad Medical City, Riyadh, Saudi Arabia. 4Clinical Geneticist, Pediatric Subspecialties Department, Children's Specialized Hospital, King Fahad Medical City, Riyadh, Saudi Arabia. 5 Research Center, King Fahad Medical City, Riyadh , Saudi Arabia What is already known on this topic ? Autosomal-recessive pseudohypoaldosteronism type 1 (PHA1) is a rare genetic disorder caused by different variations in the ENaC subunit genes. Most of these variations appear in SCNN1A mainly in exon eight, which encodes for the alpha subunit of the epithelial sodium channel ENaC. Variations are nonsense, single-base deletions or insertions, or splice site variations, leading to mRNA and proteins of abnormal length. In addition, a few new missense variations have been reported. What this study adds ? We report a novel mutation [ c.729_730delAG (p.Val245Glyfs*65) ] in the exon 4 of the SCNN1A gene In case of autosomal recessive pseudohypoaldosteronism type 1. Patient with PHA1 requires early recognition, proper treatment, and close follow-up. Parents are advised to seek genetic counseling and plan future pregnancies. proof Abstract Pseudohypoaldosteronism type 1 (PHA1) is an autosomal-recessive disorder characterized by defective regulation of body sodium levels. -

Inheriting Genetic Conditions
Help Me Understand Genetics Inheriting Genetic Conditions Reprinted from MedlinePlus Genetics U.S. National Library of Medicine National Institutes of Health Department of Health & Human Services Table of Contents 1 What does it mean if a disorder seems to run in my family? 1 2 Why is it important to know my family health history? 4 3 What are the different ways a genetic condition can be inherited? 6 4 If a genetic disorder runs in my family, what are the chances that my children will have the condition? 15 5 What are reduced penetrance and variable expressivity? 18 6 What do geneticists mean by anticipation? 19 7 What are genomic imprinting and uniparental disomy? 20 8 Are chromosomal disorders inherited? 22 9 Why are some genetic conditions more common in particular ethnic groups? 23 10 What is heritability? 24 Reprinted from MedlinePlus Genetics (https://medlineplus.gov/genetics/) i Inheriting Genetic Conditions 1 What does it mean if a disorder seems to run in my family? A particular disorder might be described as “running in a family” if more than one person in the family has the condition. Some disorders that affect multiple family members are caused by gene variants (also known as mutations), which can be inherited (passed down from parent to child). Other conditions that appear to run in families are not causedby variants in single genes. Instead, environmental factors such as dietary habits, pollutants, or a combination of genetic and environmental factors are responsible for these disorders. It is not always easy to determine whether a condition in a family is inherited. -

Restoration of Fertility by Gonadotropin Replacement in a Man With
J Rohayem and others Fertility in hypogonadotropic 170:4 K11–K17 Case Report CAH with TARTs Restoration of fertility by gonadotropin replacement in a man with hypogonadotropic azoospermia and testicular adrenal rest tumors due to untreated simple virilizing congenital adrenal hyperplasia Julia Rohayem1, Frank Tu¨ ttelmann2, Con Mallidis3, Eberhard Nieschlag1,4, Sabine Kliesch1 and Michael Zitzmann1 Correspondence should be addressed 1Center of Reproductive Medicine and Andrology, Clinical Andrology, University of Muenster, Albert-Schweitzer- to J Rohayem Campus 1, Building D11, D-48149 Muenster, Germany, 2Institute of Human Genetics and 3Institute of Reproductive Email and Regenerative Biology, Center of Reproductive Medicine and Andrology, University of Muenster, Muenster, Julia.Rohayem@ Germany and 4Center of Excellence in Genomic Medicine Research, King Abdulaziz University, Jeddah, Saudi Arabia ukmuenster.de Abstract Context: Classical congenital adrenal hyperplasia (CAH), a genetic disorder characterized by 21-hydroxylase deficiency, impairs male fertility, if insufficiently treated. Patient: A 30-year-old male was referred to our clinic for endocrine and fertility assessment after undergoing unilateral orchiectomy for a suspected testicular tumor. Histopathological evaluation of the removed testis revealed atrophy and testicular adrenal rest tumors (TARTs) and raised the suspicion of underlying CAH. The remaining testis was also atrophic (5 ml) with minor TARTs. Serum 17-hydroxyprogesterone levels were elevated, cortisol levels were at the lower limit of normal range, and gonadotropins at prepubertal levels, but serum testosterone levels were within the normal adult range. Semen analysis revealed azoospermia. CAH was confirmed by a homozygous mutation g.655A/COG (IVS2-13A/COG) in European Journal of Endocrinology CYP21A2. Hydrocortisone (24 mg/m2) administered to suppress ACTH and adrenal androgen overproduction unmasked deficient testicular testosterone production. -

A New Age in the Genetics of Deafness
, September/October 1999. Vol. 1 . No. 6 invited review/cme article A new age in the genetics of deafness Heidi L. Rehtu, MMSC' ot~dCytltllia C. Morton, PAD' Over the last several years, an understanding of the genetics SYNDROMIC AND NONSYNDROMIC DEAFNESS of hearing and deafness has grown exponentially. This progress , The prevalence of severe to profound bilateral congenital has been fueled by fast-paced developments in gene mapping hearing loss is estimated at 1 in 1000 births? When milder I and discovery, leading to the recent cloning and characteriza- forms of permanent hearing impairment at birth are included, tion of many genes critical in the biology of hearing. Among this estimate increases four-fold. Congenital deafness refers to the most significant advances, especially from a clinical per- deafness present at birth, though the cause of such hearing spective, is the discovery that up to 50% of nonsyndromic re- impairment may be genetic (hereditary) or environmental (ac- cessive deafness is caused by mutations in the GIB2 gene, which quired). Various studies estimate that approximately one-half encodes the connexin 26 protein (Cx26).I.' The clinical use of of the cases of congenital deafness are due to genetic factor^,^ screening for mutations in Cx26 and other genes involved in and these factors can be further subdivided by mode of inher- hearing impairment is still a new concept and not widely avail- itance. Approximately 77% of cases of hereditary deafness are able. However, the eventual availability of an array of genetic autosomal recessive, 22% are autosomal dominant, and 1% are tests is likely to have a major impact on the medical decisions X-linked.-l In addition, a small fraction of hereditary deafness made by hearing-impaired patients, their families, and their (< 1%) represents those families with mitochondria1 inheri- physicians. -

Waardenburg Syndrome: Case Series
International Journal of Otorhinolaryngology and Head and Neck Surgery Sachdeva S et al. Int J Otorhinolaryngol Head Neck Surg. 2021 Apr;7(4):668-671 http://www.ijorl.com pISSN 2454-5929 | eISSN 2454-5937 DOI: https://dx.doi.org/10.18203/issn.2454-5929.ijohns20211191 Case Series Waardenburg syndrome: case series Sheenu Sachdeva*, Varunkumar Jayakumar, Shubhlaxmi Atmaram Jaiswal Department of Otorhinolaryngology, Dr. V. M. G. M. C Solapur, Maharashtra, India Received: 13 January 2021 Revised: 16 February 2021 Accepted: 06 March 2021 *Correspondence: Dr. Sheenu Sachdeva, E-mail: [email protected] Copyright: © the author(s), publisher and licensee Medip Academy. This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT Waardenburg syndrome is a rare genetic disorder of neural crest cell development with incidence of 1:42000 to 1:50,000. The syndrome is not completely expressed and hence adds to its hetergenecity with symptoms varying from one type of syndrome to another and from one patient to another. Unilateral heterochromia that manifests in some people is associated with Waardenburg syndrome and Parry-Romberg syndrome. This is a case series of four cases with features of Waardenburg syndrome with variable presentations and familial inheritance. Keywords: Autosomal dominant deafness, Heterochromia, Pigmentation anomalies, Waardenburg syndrome INTRODUCTION normal. Her mother also had similar hypertelorism with normal hearing (Figure 2). Also there is history suggestive Waardenburg syndrome (WS) is a rare genetic disorder of of premature graying of hair in her grandmother since the neural crest development characterized by varying degrees age of 15 years. -

Hearing Loss in Waardenburg Syndrome: a Systematic Review
Clin Genet 2016: 89: 416–425 © 2015 John Wiley & Sons A/S. Printed in Singapore. All rights reserved Published by John Wiley & Sons Ltd CLINICAL GENETICS doi: 10.1111/cge.12631 Review Hearing loss in Waardenburg syndrome: a systematic review Song J., Feng Y., Acke F.R., Coucke P., Vleminckx K., Dhooge I.J. Hearing J. Songa,Y.Fenga, F.R. Ackeb, loss in Waardenburg syndrome: a systematic review. P. Couckec,K.Vleminckxc,d Clin Genet 2016: 89: 416–425. © John Wiley & Sons A/S. Published by and I.J. Dhoogeb John Wiley & Sons Ltd, 2015 aDepartment of Otolaryngology, Xiangya Waardenburg syndrome (WS) is a rare genetic disorder characterized by Hospital, Central South University, Changsha, People’s Republic of China, hearing loss (HL) and pigment disturbances of hair, skin and iris. b Classifications exist based on phenotype and genotype. The auditory Department of Otorhinolaryngology, cDepartment of Medical Genetics, Ghent phenotype is inconsistently reported among the different Waardenburg types University/Ghent University Hospital, and causal genes, urging the need for an up-to-date literature overview on Ghent, Belgium, and dDepartment for this particular topic. We performed a systematic review in search for articles Biomedical Molecular Biology, Ghent describing auditory features in WS patients along with the associated University, Ghent, Belgium genotype. Prevalences of HL were calculated and correlated with the different types and genes of WS. Seventy-three articles were included, describing 417 individual patients. HL was found in 71.0% and was Key words: genotype – hearing loss – predominantly bilateral and sensorineural. Prevalence of HL among the inner ear malformation – phenotype – different clinical types significantly differed (WS1: 52.3%, WS2: 91.6%, Waardenburg syndrome WS3: 57.1%, WS4: 83.5%). -
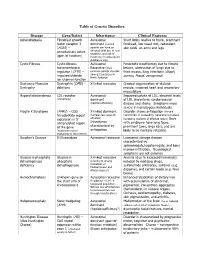
Table of Genetic Disorders Disease Gene/Defect Inheritance Clinical
Table of Genetic Disorders Disease Gene/Defect Inheritance Clinical Features Achondroplasia Fibroblast growth Autosomal Short limbs relative to trunk, prominent factor receptor 3 dominant (normal forehead, low nasal root, redundant (FGR3) – parents can have an skin folds on arms and legs constitutively active affected child due to new mutation, and risk of (gain of function) recurrence in subsequent children is low) Cystic Fibrosis Cystic fibrosis Autosomal Pancreatic insufficiency due to fibrotic transmembrane Recessive (most lesions, obstruction of lungs due to regulator (CFTR) – common genetic disorder thick mucus, lung infections (Staph, impaired chloride among Caucasians in aureus, Pseud. aeruginosa) North America) ion channel function Duchenne Muscular Dystrophin (DMD) - X-linked recessive Gradual degeneration of skeletal Dystrophy deletions muscle, impaired heart and respiratory musculature Hypercholesterolemia LDL receptor Autosomal Impaired uptake of LDL, elevated levels (commonly) dominant of LDL cholesterol, cardiovascular (haploinsufficiency) disease and stroke. Symptoms more severe in homozygous individuals Fragile X Syndrome (FMR1) – CGG X-linked dominant Disorder shows anticipation (female trinucleotide repeat (females less severely transmitters in succeeding generations produce expansion in 5’ affected) increasing numbers of affected males) Boys untranslated region Inheritance with syndrome have long faces, of the gene characterized by prominent jaws, large ears, and are (expansion occurs anticipation likely to be mentally retarded. exclusively in the mother) Gaucher’s Disease Β-Glucosidase Autosomal recessive Lysosomal storage disease characterized by splenomegaly,hepatomegaly, and bone marrow infiltration. Neurological symptoms are not common Glucose 6-phosphate Glucose 6- X-linked recessive Anemia (due to increased hemolysis) dehydrogenase phosphate (prominent among induced by oxidizing drugs, deficiency dehydrogenase individuals of sulfonamide antibiotics, sulfones (e.g. -

Hereditary Hearing Impairment with Cutaneous Abnormalities
G C A T T A C G G C A T genes Review Hereditary Hearing Impairment with Cutaneous Abnormalities Tung-Lin Lee 1 , Pei-Hsuan Lin 2,3, Pei-Lung Chen 3,4,5,6 , Jin-Bon Hong 4,7,* and Chen-Chi Wu 2,3,5,8,* 1 Department of Medical Education, National Taiwan University Hospital, Taipei City 100, Taiwan; [email protected] 2 Department of Otolaryngology, National Taiwan University Hospital, Taipei 11556, Taiwan; [email protected] 3 Graduate Institute of Clinical Medicine, National Taiwan University College of Medicine, Taipei City 100, Taiwan; [email protected] 4 Graduate Institute of Medical Genomics and Proteomics, National Taiwan University College of Medicine, Taipei City 100, Taiwan 5 Department of Medical Genetics, National Taiwan University Hospital, Taipei 10041, Taiwan 6 Department of Internal Medicine, National Taiwan University Hospital, Taipei 10041, Taiwan 7 Department of Dermatology, National Taiwan University Hospital, Taipei City 100, Taiwan 8 Department of Medical Research, National Taiwan University Biomedical Park Hospital, Hsinchu City 300, Taiwan * Correspondence: [email protected] (J.-B.H.); [email protected] (C.-C.W.) Abstract: Syndromic hereditary hearing impairment (HHI) is a clinically and etiologically diverse condition that has a profound influence on affected individuals and their families. As cutaneous findings are more apparent than hearing-related symptoms to clinicians and, more importantly, to caregivers of affected infants and young individuals, establishing a correlation map of skin manifestations and their underlying genetic causes is key to early identification and diagnosis of syndromic HHI. In this article, we performed a comprehensive PubMed database search on syndromic HHI with cutaneous abnormalities, and reviewed a total of 260 relevant publications. -

Mutation of the KIT (Mast/Stem Cell Growth Factor Receptor
Proc. Nati. Acad. Sci. USA Vol. 88, pp. 8696-8699, October 1991 Genetics Mutation of the KIT (mast/stem cell growth factor receptor) protooncogene in human piebaldism (pigmentation disorders/white spottlng/oncogene/receptor/tyrosine kinase) LUTZ B. GIEBEL AND RICHARD A. SPRITZ* Departments of Medical Genetics and Pediatrics, 317 Laboratory of Genetics, University of Wisconsin, Madison, WI 53706 Communicated by James F. Crow, July 8, 1991 ABSTRACT Piebaldism is an autosomal dominant genetic represent a human homologue to dominant white spotting disorder characterized by congenital patches of skin and har (W) of the mouse. from which melanocytes are completely absent. A similar disorder of mouse, dominant white spotting (W), results from MATERIALS AND METHODS mutations of the c-Kit protooncogene, which encodes the re- ceptor for mast/stem cell growth factor. We identified a KIT Description ofthe Probaind. The probandt was an adult man gene mutation in a proband with classic autosomal dint with typical features of piebaldism, including nonpigmented piebaldism. This mutation results in a Gly -- Arg substitution patches on his central forehead, central chest and abdomen, at codon 664, within the tyrosine kinase domain. This substi- and arms and legs. Pigmentation of his scalp hair and facial tution was not seen in any normal individuals and was com- hair was normal, although several other family members had pletely linked to the piebald phenotype in the proband's family. white forelocks in addition to nonpigmented skin patches. Piebaldism in this family thus appears to be the human His irides and retinae were normally pigmented, and hearing homologue to dominant white spotting (W) of the mouse. -

2012 CKD Guideline
OFFICIAL JOURNAL OF THE INTERNATIONAL SOCIETY OF NEPHROLOGY KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease VOLUME 3 | ISSUE 1 | JANUARY 2013 http://www.kidney-international.org KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease KDIGO gratefully acknowledges the following consortium of sponsors that make our initiatives possible: Abbott, Amgen, Bayer Schering Pharma, Belo Foundation, Bristol-Myers Squibb, Chugai Pharmaceutical, Coca-Cola Company, Dole Food Company, Fresenius Medical Care, Genzyme, Hoffmann-LaRoche, JC Penney, Kyowa Hakko Kirin, NATCO—The Organization for Transplant Professionals, NKF-Board of Directors, Novartis, Pharmacosmos, PUMC Pharmaceutical, Robert and Jane Cizik Foundation, Shire, Takeda Pharmaceutical, Transwestern Commercial Services, Vifor Pharma, and Wyeth. Sponsorship Statement: KDIGO is supported by a consortium of sponsors and no funding is accepted for the development of specific guidelines. http://www.kidney-international.org contents & 2013 KDIGO VOL 3 | ISSUE 1 | JANUARY (1) 2013 KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease v Tables and Figures vii KDIGO Board Members viii Reference Keys x CKD Nomenclature xi Conversion Factors & HbA1c Conversion xii Abbreviations and Acronyms 1 Notice 2 Foreword 3 Work Group Membership 4 Abstract 5 Summary of Recommendation Statements 15 Introduction: The case for updating and context 19 Chapter 1: Definition, and classification -

Diagnosis, Treatment, and Outcomes in Children with Congenital Nephrogenic Diabetes Insipidus: a Pediatric Nephrology Research Consortium Study
Journal Articles 2020 Diagnosis, Treatment, and Outcomes in Children With Congenital Nephrogenic Diabetes Insipidus: A Pediatric Nephrology Research Consortium Study C. D'Alessandri-Silva M. Carpenter R. Ayoob J. Barcia A. Chishti See next page for additional authors Follow this and additional works at: https://academicworks.medicine.hofstra.edu/articles Part of the Pediatrics Commons Recommended Citation D'Alessandri-Silva C, Carpenter M, Ayoob R, Barcia J, Chishti A, Constantinescu A, Dell KM, Goodwin J, Sethna C, Mahan JD, . Diagnosis, Treatment, and Outcomes in Children With Congenital Nephrogenic Diabetes Insipidus: A Pediatric Nephrology Research Consortium Study. 2020 Jan 01; 7():Article 7841 [ p.]. Available from: https://academicworks.medicine.hofstra.edu/articles/7841. Free full text article. This Article is brought to you for free and open access by Donald and Barbara Zucker School of Medicine Academic Works. It has been accepted for inclusion in Journal Articles by an authorized administrator of Donald and Barbara Zucker School of Medicine Academic Works. For more information, please contact [email protected]. Authors C. D'Alessandri-Silva, M. Carpenter, R. Ayoob, J. Barcia, A. Chishti, A. Constantinescu, K. M. Dell, J. Goodwin, C. Sethna, J. D. Mahan, and +9 additional authors This article is available at Donald and Barbara Zucker School of Medicine Academic Works: https://academicworks.medicine.hofstra.edu/articles/7841 ORIGINAL RESEARCH published: 21 January 2020 doi: 10.3389/fped.2019.00550 Diagnosis, Treatment, and Outcomes in Children With Congenital Nephrogenic Diabetes Insipidus: A Pediatric Nephrology Research Consortium Study Cynthia D’Alessandri-Silva 1,2*, Melinda Carpenter 1,3, Rose Ayoob 4, John Barcia 5, Aftab Chishti 6, Alex Constantinescu 7, Katherine M.