DISSERTATION a SELECTION of NITRIC OXIDE-RELEASING MATERIALS INCORPORATING S- NITROSOTHIOLS Submitted by Alec Lutzke Department
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Inhaled Nitric Oxide Therapy in Adults Benedict C Creagh-Brown, Mark JD Griffiths and Timothy W Evans
Available online http://ccforum.com/content/13/3/212 Review Bench-to-bedside review: Inhaled nitric oxide therapy in adults Benedict C Creagh-Brown, Mark JD Griffiths and Timothy W Evans Unit of Critical Care, Faculty of Medicine, Imperial College, London, UK and Adult Intensive Care Unit, Royal Brompton Hospital, Sydney Street, London, SW3 6NP, UK Corresponding author: Timothy W Evans, [email protected] Published: 29 May 2009 Critical Care 2009, 13:212 (doi:10.1186/cc7734) This article is online at http://ccforum.com/content/13/3/221 © 2009 BioMed Central Ltd Abstract Administration of inhaled nitric oxide to adults Nitric oxide (NO) is an endogenous mediator of vascular tone and The licensed indication of iNO is restricted to persistent host defence. Inhaled nitric oxide (iNO) results in preferential pulmonary hypertension in neonates, yet most iNO is pulmonary vasodilatation and lowers pulmonary vascular resis- administered for unlicensed indications. Pharmaceutical iNO tance. The route of administration delivers NO selectively to is available at a very high cost, and in light of this and ventilated lung units so that its effect augments that of hypoxic concerns over potential adverse effects of iNO, international pulmonary vasoconstriction and improves oxygenation. This guidelines have been developed. An advisory board under the ‘Bench-to-bedside’ review focuses on the mechanisms of action of iNO and its clinical applications, with emphasis on acute lung injury auspices of the European Society of Intensive Care Medicine and the acute respiratory distress syndrome. Developments in our and the European Association of Cardiothoracic Anaes- understanding of the cellular and molecular actions of NO may thesiologists published its recommendations in 2005 [1]. -

Aldrich Raman
Aldrich Raman Library Listing – 14,033 spectra This library represents the most comprehensive collection of FT-Raman spectral references available. It contains many common chemicals found in the Aldrich Handbook of Fine Chemicals. To create the Aldrich Raman Condensed Phase Library, 14,033 compounds found in the Aldrich Collection of FT-IR Spectra Edition II Library were excited with an Nd:YVO4 laser (1064 nm) using laser powers between 400 - 600 mW, measured at the sample. A Thermo FT-Raman spectrometer (with a Ge detector) was used to collect the Raman spectra. The spectra were saved in Raman Shift format. Aldrich Raman Index Compound Name Index Compound Name 4803 ((1R)-(ENDO,ANTI))-(+)-3- 4246 (+)-3-ISOPROPYL-7A- BROMOCAMPHOR-8- SULFONIC METHYLTETRAHYDRO- ACID, AMMONIUM SALT PYRROLO(2,1-B)OXAZOL-5(6H)- 2207 ((1R)-ENDO)-(+)-3- ONE, BROMOCAMPHOR, 98% 12568 (+)-4-CHOLESTEN-3-ONE, 98% 4804 ((1S)-(ENDO,ANTI))-(-)-3- 3774 (+)-5,6-O-CYCLOHEXYLIDENE-L- BROMOCAMPHOR-8- SULFONIC ASCORBIC ACID, 98% ACID, AMMONIUM SALT 11632 (+)-5-BROMO-2'-DEOXYURIDINE, 2208 ((1S)-ENDO)-(-)-3- 97% BROMOCAMPHOR, 98% 11634 (+)-5-FLUORODEOXYURIDINE, 769 ((1S)-ENDO)-(-)-BORNEOL, 99% 98+% 13454 ((2S,3S)-(+)- 11633 (+)-5-IODO-2'-DEOXYURIDINE, 98% BIS(DIPHENYLPHOSPHINO)- 4228 (+)-6-AMINOPENICILLANIC ACID, BUTANE)(N3-ALLYL)PD(II) CL04, 96% 97 8167 (+)-6-METHOXY-ALPHA-METHYL- 10297 ((3- 2- NAPHTHALENEACETIC ACID, DIMETHYLAMINO)PROPYL)TRIPH 98% ENYL- PHOSPHONIUM BROMIDE, 12586 (+)-ANDROSTA-1,4-DIENE-3,17- 99% DIONE, 98% 13458 ((R)-(+)-2,2'- 963 (+)-ARABINOGALACTAN BIS(DIPHENYLPHOSPHINO)-1,1'- -

Airway Nitrite Is Increased in Extremely Preterm Infants with Bronchopulmonary Dysplasia Samuel J
Gentle et al. Respiratory Research (2020) 21:244 https://doi.org/10.1186/s12931-020-01508-8 LETTER TO THE EDITOR Open Access Airway nitrite is increased in extremely preterm infants with bronchopulmonary dysplasia Samuel J. Gentle1* , Amelia Freeman1, Rakesh P. Patel2, Namasivayam Ambalavanan1 and Charitharth V. Lal1 Abstract Rationale: Bronchopulmonary dysplasia (BPD) is the most common complication of prematurity and significantly contributes to mortality and morbidity with few predictive biomarkers. Given that nitrites have been implicated in pathways associated with lung disease, we hypothesized that nitrite levels would be altered in the airways of premature infants diagnosed with BPD. Methods: This was a prospective cohort study of extremely low birth infants (< 28 weeks’ gestation) at the University of Alabama at Birmingham. Nitrite levels from tracheal aspirates (TAs) were compared between intubated and ventilated infants with BPD and gestation matched full term (FT) controls. TA derived nitrite levels from day one after birth were also compared between preterm infants who did and did not develop BPD. Results: Infants with BPD were found to have significantly elevated nitrite levels in their tracheal aspirates compared to gestation matched FT controls (p < 0.05). There was a trend for increased nitrite levels on postnatal day one in infants that developed BPD compared to infants that did not develop BPD (p = 0.05). Conclusions: In conclusion, nitrite levels are significantly increased in airways of infants with BPD. Data from a larger cohort are needed to further support the utility of nitrite for BPD prediction. Trial registration: Not applicable. Keywords: Bronchopulmonary dysplasia, Preterm infants, Nitrite, Nitric oxide Introduction Nitric oxide a signaling molecule produced throughout Bronchopulmonary dysplasia (BPD) is a common morbid- the airway, has many physiologic roles in the lung in- ity of preterm infants for which few biomarkers exist. -

Prohibited and Restricted Chemical List
School Emergency Response Plan and Management Guide Prohibited and Restricted Chemical List PROHIBITED AND RESTRICTED CHEMICAL LIST Introduction After incidents of laboratory chemical contamination at several schools, DCPS, The American Association for the Advancement of Science (AAAS) and DC Fire and Emergency Management Services developed an aggressive program for chemical control to eliminate student and staff exposure to potential hazardous chemicals. Based upon this program, all principals are required to conduct a complete yearly inventory of all chemicals located at each school building to identify for the removal and disposal of any prohibited/banned chemicals. Prohibited chemicals are those that pose an inherent, immediate, and potentially life- threatening risk, injury, or impairment due to toxicity or other chemical properties to students, staff, or other occupants of the school. These chemicals are prohibited from use and/or storage at the school, and the school is prohibited from purchasing or accepting donations of such chemicals. Restricted chemicals are chemicals that are restricted by use and/or quantities. If restricted chemicals are present at the school, each storage location must be addressed in the school's written emergency plan. Also, plan maps must clearly denote the storage locations of these chemicals. Restricted chemicals—demonstration use only are a subclass in the Restricted chemicals list that are limited to instructor demonstration. Students may not participate in handling or preparation of restricted chemicals as part of a demonstration. If Restricted chemicals—demonstration use only are present at the school, each storage location must be addressed in the school's written emergency plan. Section 7: Appendices – October 2009 37 School Emergency Response Plan and Management Guide Prohibited and Restricted Chemical List Following is a table of chemicals that are Prohibited—banned, Restricted—academic curriculum use, and Restricted—demonstration use only. -

Potentially Explosive Chemicals*
Potentially Explosive Chemicals* Chemical Name CAS # Not 1,1’-Diazoaminonaphthalene Assigned 1,1-Dinitroethane 000600-40-8 1,2,4-Butanetriol trinitrate 006659-60-5 1,2-Diazidoethane 000629-13-0 1,3,5-trimethyl-2,4,6-trinitrobenzene 000602-96-0 1,3-Diazopropane 005239-06-5 Not 1,3-Dinitro-4,5-dinitrosobenzene Assigned Not 1,3-dinitro-5,5-dimethyl hydantoin Assigned Not 1,4-Dinitro-1,1,4,4-tetramethylolbutanetetranitrate Assigned Not 1,7-Octadiene-3,5-Diyne-1,8-Dimethoxy-9-Octadecynoic acid Assigned 1,8 –dihydroxy 2,4,5,7-tetranitroanthraquinone 000517-92-0 Not 1,9-Dinitroxy pentamethylene-2,4,6,8-tetramine Assigned 1-Bromo-3-nitrobenzene 000585-79-5 Not 2,2',4,4',6,6'-Hexanitro-3,3'-dihydroxyazobenzene Assigned 2,2-di-(4,4,-di-tert-butylperoxycyclohexyl)propane 001705-60-8 2,2-Dinitrostilbene 006275-02-1 2,3,4,6- tetranitrophenol 000641-16-7 Not 2,3,4,6-tetranitrophenyl methyl nitramine Assigned Not 2,3,4,6-tetranitrophenyl nitramine Assigned Not 2,3,5,6- tetranitroso nitrobenzene Assigned Not 2,3,5,6- tetranitroso-1,4-dinitrobenzene Assigned 2,4,6-Trinitro-1,3,5-triazo benzene 029306-57-8 Not 2,4,6-trinitro-1,3-diazabenzene Assigned Not 2,4,6-Trinitrophenyl trimethylol methyl nitramine trinitrate Assigned Not 2,4,6-Trinitroso-3-methyl nitraminoanisole Assigned 2,4-Dinitro-1,3,5-trimethyl-benzene 000608-50-4 2,4-Dinitrophenylhydrazine 000119-26-6 2,4-Dinitroresorcinol 000519-44-8 2,5-dimethyl-2,5-diydroperoxy hexane 2-Nitro-2-methylpropanol nitrate 024884-69-3 3,5-Dinitrosalicylic acid 000609-99-4 Not 3-Azido-1,2-propylene glycol dinitrate -

STUDIES on the SYNTHESIS and REACTIVITY of FIVE-MEMBERED HETEROCYCLIC DIAZONIUM CATIONS by WILLIAM NICOL DUFF, B.Sc. Thesis Pres
STUDIES ON THE SYNTHESIS AND REACTIVITY OF FIVE-MEMBERED HETEROCYCLIC DIAZONIUM CATIONS by WILLIAM NICOL DUFF, B.Sc. Thesis presented for the degree of Doctor of Philosophy University of Edinburgh 1981 Ez 08 rn Ze 0 8 X1 DECLARATION I declare that this thesis is my own composition, that the work of which it is a record has been carried out by myself and that it has not been submitted in any previous application for a Higher Degree. The thesis describes the results of research carried out in the Department of Chemistry, University of Edinburgh, under the supervision of Dr. G. Tennant between October 1976 and September 1979. ACKNOWLEDGEMENTS I should like to place on record my appreciation of the guidance and encouragement provided by my supervisor Dr. G. Tennant. I am grateful for the patience he displayed over the years of our association. I should like to thank the Science Research Council in collaboration with I. C. I. Organics Division, Blackley, Manchester for the award of a C.A.S.E. studentship over three years. lam also grateful to the University of Edinburgh for the provision of laboratory and library facilities. I am further indebted to the technical staff of the Department of Chemistry, University of Edinburgh for their help and assistance. I should also like to acknowledge Dr. D. B. Baird of I.C.I. Organics Division for his many helpful suggestions and his supervision during my stay in Blackley. I should like to thank Mrs. C. Ranken for her care and patience typing this manuscript. POSTGRADUATE LECTURE COURSES ATTENDED BETWEEN OCTOBER 1976 AND SEPTEMBER 1979 "Optical Properties of Transition Metal Complexes, 11 Dr. -

United States Patent (10 ) Patent No.: US 10,555,968 B2 Lundberg Et Al
US010555968B2 United States Patent (10 ) Patent No.: US 10,555,968 B2 Lundberg et al. (45 ) Date of Patent: Feb. 11 , 2020 (54 ) PERFORMANCE ENHANCING A61K 9/0056 (2013.01 ) ; A61K 31/375 COMPOSITION AND USE THEREOF ( 2013.01 ) ; A61K 36/21 (2013.01 ) ; A61K 45/06 ( 2013.01 ) ; A23V 2002/00 ( 2013.01 ) ; AIK ( 71 ) Applicant : HeartBeet Ltd., Ipswich (GB ) 36/185 ( 2013.01) ; COID 9/00 (2013.01 ) (72 ) Inventors : Jon Lundberg , Djursholm (SE ); Eddie (58 ) Field of Classification Search Weitzberg , Stockholm (SE ) None See application file for complete search history . ( * ) Notice : Subject to any disclaimer , the term of this patent is extended or adjusted under 35 ( 56 ) References Cited U.S.C. 154 ( b ) by 0 days. U.S. PATENT DOCUMENTS ( 21) Appl. No .: 14 /830,937 4,868,179 A 9/1989 Cohn ( 22 ) Filed : Aug. 20 , 2015 5,476,847 A 12/1995 McKittrick et al . (Continued ) (65 ) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2015/0352147 A1 Dec. 10 , 2015 EP 0511587 A1 11/1992 Related U.S. Application Data KR 2002/0057695 A 7/2002 (63 ) Continuation of application No. 12 /528,798 , filed as (Continued ) application No. PCT /SE2008 / 050211 on Feb. 26 , 2008 , now Pat . No. 9,180,140 . OTHER PUBLICATIONS ( 30 ) Foreign Application Priority Data Gladwyn et al. , Nature Chemical Biology, vol. 1 No. 6 Nov. 2005 , pp . 308-314 ( 2005 ) . * Feb. 26 , 2007 ( SE ) 0700520 Mar. 22 , 2007 (SE ) 0700729 ( Continued ) (51 ) Int. Ci. Primary Examiner Bethany P Barham A61K 33/00 ( 2006.01 ) Assistant Examiner Barbara S Frazier A61K 9/00 ( 2006.01 ) ( 74 ) Attorney , Agent, or Firm - Booth Udall Fuller , PLC A61K 31/375 ( 2006.01) A61K 36/185 ( 2006.01 ) (57 ) ABSTRACT A61K 36/21 ( 2006.01 ) The performance of a mammal, manifested as a reduced A61K 45/06 (2006.01 ) oxygen uptake (Vo ) during physical exercise , can be A23L 2/52 ( 2006.01 ) enhanced by administering to said mammal a non - toxic (Continued ) amount of nitrate and / or nitrite . -

List of Reactive Chemicals
LIST OF REACTIVE CHEMICALS Chemical Prefix Chemical Name Reactive Reactive Reactive CAS# Chemical Chemical Chemical Stimulus 1 Stimulus 2 Stimulus 3 111-90-0 "CARBITOL" SOLVENT D 111-15-9 "CELLOSOLVE" ACETATE D 110-80-5 "CELLOSOLVE" SOLVENT D 2- (2,4,6-TRINITROPHENYL)ETHYL ACETATE (1% IN ACETONE & BENZENE S 12427-38-2 AAMANGAN W 88-85-7 AATOX S 40487-42-1 AC 92553 S 105-57-7 ACETAL D 75-07-0 ACETALDEHYDE D 105-57-7 ACETALDEHYDE, DIETHYL ACETAL D 108-05-4 ACETIC ACID ETHENYL ESTER D 108-05-4 ACETIC ACID VINYL ESTER D 75-07-0 ACETIC ALDEHYDE D 101-25-7 ACETO DNPT T 126-84-1 ACETONE DIETHYL ACETAL D 108-05-4 ACETOXYETHYLENE D 108-05-4 1- ACETOXYETHYLENE D 37187-22-7 ACETYL ACETONE PEROXIDE, <=32% AS A PASTE T 37187-22-7 ACETYL ACETONE PEROXIDE, <=42% T 37187-22-7 ACETYL ACETONE PEROXIDE, >42% T S 644-31-5 ACETYL BENZOYL PEROXIDE (SOLID OR MORE THAN 45% IN SOLUTION) T S 644-31-5 ACETYL BENZOYL PEROXIDE, <=45% T 506-96-7 ACETYL BROMIDE W 75-36-5 ACETYL CHLORIDE W ACETYL CYCLOHEXANE SULFONYL PEROXIDE (>82% WITH <12% WATER) T S 3179-56-4 ACETYL CYCLOHEXANE SULFONYL PEROXIDE, <=32% T 3179-56-4 ACETYL CYCLOHEXANE SULFONYL PEROXIDE, <=82% T 674-82-8 ACETYL KETENE (POISON INHALATION HAZARD) D 110-22-5 ACETYL PEROXIDE, <=27% T 110-22-5 ACETYL PEROXIDE, SOLID, OR MORE THAN 27% IN SOLUTION T S 927-86-6 ACETYLCHOLINE PERCHLORATE O S 74-86-2 ACETYLENE D 74-86-2 ACETYLENE (LIQUID) D ACETYLENE SILVER NITRATE D 107-02-08 ACRALDEHYDE (POISON INHALATION HAZARD) D 79-10-7 ACROLEIC ACID D 107-02-08 ACROLEIN, INHIBITED (POISON INHALATION HAZARD) D 107-02-08 ACRYLALDEHYDE (POISON INHALATION HAZARD) D 79-10-7 ACRYLIC ACID D 141-32-2 ACRYLIC ACID BUTYL ESTER D 140-88-5 ACRYLIC ACID ETHYL ESTER D 96-33-3 ACRYLIC ACID METHYL ESTER D Stimulus - Stimuli is the thermal, physical or chemical input needed to induce a hazardous reaction. -

W O 2016/090373 Al 9 June 2016 (09.06.2016) W I PO I P CT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date W O 2016/090373 Al 9 June 2016 (09.06.2016) W I PO I P CT (51) International Patent Classification: (81) Designated States (unless otherwise indicated,for every A61K31/417 (2006.01) GO1N 33/573 (2006.01) kind of national protection available): AE, AG, AL, AM, A61K31/4172 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, ) .a BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, (21) International Application Number: DO, DZ, EC, EE, EG, ES, Fl, GB, GD, GE, GH, GM, GT, PCT/US2015/064308 HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (22) International Filing Date: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, 7 December 2015 (07.12.2015) MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (25) Filing Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (26) Publication Language: English TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: (84) Designated States (unless otherwise indicated,for every 62/088,002 5 December 2014 (05.12.2014) US kind of regional protection available): ARIPO (BW, GH, GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, (71) Applicant: CASE WESTERN RESERVE UNIVER- TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, SITY [US/US]; 10900 Euclid Avenue, Cleveland, Ohio TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, 44106 (US). -
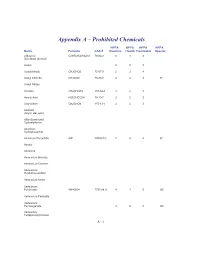
Appendix a – Prohibited Chemicals
Appendix A – Prohibited Chemicals NFPA NFPA NFPA NFPA Name Formula CAS #Reactive Health Flammable Special 2-Butanol C2H5CH(OH)CH3 78-92-2 0 1 3 (Sec-Butyl Alcohol) Acetal 023 Acetaldehyde CH3CHO5 75-07-0 2 3 4 Acetyl Chloride CH3COCl 75-36-5 2 3 3 W Acetyl Nitrate Acrolein CH2CHCHO 107-02-8 3 4 3 Acrylic Acid H2CCHCO2H 79-10-7 2 2 2 Acrylonitrile CH2CHCN 107-13-1 2 4 3 Alcohols (Allylic, Benzylic) Alkly-Substituted Cycloaliphatics Aluminum Hydrophosphide Aluminum Phosphide AlP 20859-73- 2 4 4 W Amatol Ammonal Ammonium Bromate Ammonium Chlorate Ammonium Hexanitrocobaltate Ammonium Nitrite Ammonium Perchlorate NH4ClO4 7790-98-9 4 1 0 OX Ammonium Periodate Ammonium Permanganate 300 OX Ammonium Tetraperoxychromate A - 1 Appendix A – Prohibited Chemicals NFPA NFPA NFPA NFPA Name Formula CAS #Reactive Health Flammable Special Antimony Compounds Arsenic And Arsenic Compounds Azides Azidocarbonyl Guanidine Barium Ba 2 2 1 W Barium Chlorate Ba(ClO3)2*H2O 13477-00- 1 2 0 OX Barium Oxide (Anhydrous) BaO 1304-28-5 2 3 0 Barium Peroxide BaO2 1304-29-6 0 1 0 OX Benzene C6H6 71-43-2 0 2 3 Benzene Diazonium Chloride Benzotriazole C6H5N3 95-14-7 0 2 1 Benzoyl Peroxide (C6H5CO)2O2 94-36-0 4 1 4 OX Benzyl Alcohol C6H5CH2OH 100-51-6 0 2 1 Bismuth Nitrate Bi(NO3)3*5H2O 10035-06- 3 1 0 OX Borane,Boranes, Diboranes Boron Tribromide 230 W Boron Trifluoride 140 Bromine Pentafluoride Brf5 7789-30-2 3 4 0 W,O Bromine Trifluoride 3 4 0 W,O Butadiene C4H6/CH2=(CH)2=CH 106-99-0 0 2 4 Butenetroil Trinitrate Cadmium and Cadmium Compounds Calcium Nitrate, Anhydrous Ca(NO3)2 -

ATSDR Case Studies in Environmental Medicine Nitrate/Nitrite Toxicity
ATSDR Case Studies in Environmental Medicine Nitrate/Nitrite Toxicity Agency for Toxic Substances and Disease Registry Case Studies in Environmental Medicine (CSEM) Nitrate/Nitrite Toxicity Course: WB2342 CE Original Date: December 5, 2013 CE Renewal Date: December 5, 2015 CE Expiration Date: December 5, 2017 Key • Nitrate toxicity is a preventable cause of Concepts methemoglobinemia. • Infants younger than 4 months of age are at particular risk of nitrate toxicity from contaminated well water. • The widespread use of nitrate fertilizers increases the risk of well-water contamination in rural areas. About This and This educational case study document is one in a series of Other Case self-instructional modules designed to increase the Studies in primary care provider’s knowledge of hazardous Environmental substances in the environment and to promote the Medicine adoption of medical practices that aid in the evaluation and care of potentially exposed patients. The complete series of Case Studies in Environmental Medicine is located on the ATSDR Web site at URL: http://www.atsdr.cdc.gov/csem/csem.html In addition, the downloadable PDF version of this educational series and other environmental medicine materials provides content in an electronic, printable format. Acknowledgements We gratefully acknowledge the work of the medical writers, editors, and reviewers in producing this educational resource. Contributors to this version of the Case Study in Environmental Medicine are listed below. Please Note: Each content expert for this case study has indicated that there is no conflict of interest that would bias the case study content. CDC/ATSDR Author(s): Kim Gehle MD, MPH CDC/ATSDR Planners: Charlton Coles, Ph.D.; Kimberly Gehle, MD; Sharon L. -

(12) United States Patent (10) Patent No.: US 7,858,378 B2 Yabuki Et Al
US007858378B2 (12) United States Patent (10) Patent No.: US 7,858,378 B2 Yabuki et al. (45) Date of Patent: Dec. 28, 2010 (54) METHOD FOR THE DETERMINATION OF Cysteine Precursors by a C-S Lyase Isolated from Axilla bacteria, BODY ODOR Chemistry & Biodiversity, 2004, vol. 1, pp. 1058-1067.* Hendriks et al., Feline: A Urinary Amino Acid of Felidae, Comp. (75) Inventors: Masayuki Yabuki, Sumida-ku (JP); Biochem. Physiol., 1995, vol. 1128, pp. 581-588.* Yoshihiro Hasegawa, Sumida-ku (JP); Wouter H. Hendriks, et al., “Felinine: A Urinary Amino Acid of Shun Nakamura, Haga-gun (JP) Felidale'. Comp. Biochem. Physiol...vol. 112B, pp. 581–588, 1995. (73) Assignee: Kao Corporation, Tokyo (JP) Natsch, Andreas et al., Identification of Odoriferous Sulfanylalkanois in Human Azilla Secretions and Their Formation through Cleavage of (*) Notice: Subject to any disclaimer, the term of this Cysteine Precursors by a C-S Lyase Isolated from Axilla bacteria, patent is extended or adjusted under 35 Chemistry & Biodiversity, 2004, vol. 1, pp. 1058-1067. U.S.C. 154(b) by 0 days. Starkenmann, Christian et al., Identificiation of the Precursor of (S)- 3-Menthyl-3-sulfanylhexan-1-ol, the Sulfury Malodor of Human (21) Appl. No.: 12/537,034 Axilla Sweat, Chemistry & Biodiversity, 2005, vol. 2, pp. 705-716. (22) Filed: Aug. 6, 2009 * cited by examiner (65) Prior Publication Data Primary Examiner Walter D Griffin Assistant Examiner Christine T. Mui US 201O/OO35355A1 Feb. 11, 2010 (74) Attorney, Agent, or Firm Oblon, Spivak, McClelland, Maier & Neustadt, L.L.P. Related U.S. Application Data (62) Division of application No.