A Modular Framework for the Development of Targeted Covid-19 Blood
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
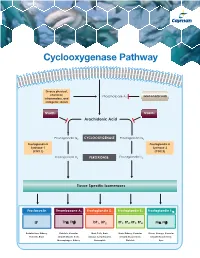
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally. -

Japanese Guidelines for Adult Asthma 2020*
Allergology International xxx (xxxx) xxx Contents lists available at ScienceDirect Allergology International journal homepage: http://www.elsevier.com/locate/alit Invited Review Article Japanese guidelines for adult asthma 2020* * Yoichi Nakamura a, , Jun Tamaoki b, Hiroyuki Nagase c, Masao Yamaguchi d, Takahiko Horiguchi e, Soichiro Hozawa f, Masakazu Ichinose g, Takashi Iwanaga h, Rieko Kondo e, Makoto Nagata i, Akihito Yokoyama j, Yuji Tohda h, The Japanese Society of Allergology a Medical Center for Allergic and Immune Diseases, Yokohama City Minato Red Cross Hospital, Yokohama, Japan b First Department of Medicine, Tokyo Women's Medical University, Tokyo, Japan c Division of Respiratory Medicine and Allergology, Department of Medicine, Teikyo University School of Medicine, Tokyo, Japan d Third Department of Medicine, Teikyo University Chiba Medical Center, Chiba, Japan e Department of Respiratory Medicine, Fujita Health University Bantane Hospital, Nagoya, Japan f Hiroshima Allergy and Respiratory Clinic, Hiroshima, Japan g Department of Respiratory Medicine, Tohoku University Graduate School of Medicine, Sendai, Japan h Department of Respiratory Medicine and Allergology, Kinki University Faculty of Medicine, Osaka, Japan i Department of Respiratory Medicine, Saitama Medical University Hospital, Saitama, Japan j Department of Hematology and Respiratory Medicine, Kochi University, Kochi, Japan article info abstract Article history: Bronchial asthma is characterized by chronic airway inflammation, which manifests clinically as variable Received 9 June 2020 airway narrowing (wheezes and dyspnea) and cough. Long-standing asthma may induce airway Available online xxx remodeling and become intractable. The prevalence of asthma has increased; however, the number of patients who die from it has decreased (1.3 per 100,000 patients in 2018). -

Role of Thromboxane Receptor-Alpha in Prostate Cancer Progression Prasanna Ekambaram Wayne State University
Wayne State University Wayne State University Dissertations 1-1-2012 Role Of Thromboxane Receptor-Alpha In Prostate Cancer Progression Prasanna Ekambaram Wayne State University, Follow this and additional works at: http://digitalcommons.wayne.edu/oa_dissertations Recommended Citation Ekambaram, Prasanna, "Role Of Thromboxane Receptor-Alpha In Prostate Cancer Progression" (2012). Wayne State University Dissertations. Paper 591. This Open Access Dissertation is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Dissertations by an authorized administrator of DigitalCommons@WayneState. ROLE OF THROMBOXANE RECEPTOR-ALPHA IN PROSTATE CANCER PROGRESSION by PRASANNA EKAMBARAM DISSERTATION Submitted to the Graduate School of Wayne State University, Detroit, Michigan in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY 2012 MAJOR: CANCER BIOLOGY Approved by: ______________________________ Advisor Date ______________________________ ______________________________ ______________________________ ______________________________ © COPYRIGHT BY PRASANNA EKAMBARAM 2012 All rights reserved DEDICATION I would like to dedicate this dissertation to my wife, Arulselvi and my kids Tejas and Abhi for standing strong by my side at good times and difficult times in life and helping me work hard toward achieving my goal of being a research scientist; To my parents for their blessings, and prayers and for helping to come to Unites States to complete my graduate studies. ii ACKNOWLEDGMENTS Foremost, I would like to extend my gratitude to my mentor, Dr. Honn for giving me this wonderful opportunity to work in his laboratory and to be my mentor for my PhD program. I would like to thank him for his immense patience, the support and guidance through the years. -

Pharmaceutical Sciences
IAJPS 2018, 05 (01), 33-36 Naveen Kumar G.S et al ISSN 2349-7750 CODEN [USA]: IAJPBB ISSN: 2349-7750 INDO AMERICAN JOURNAL OF PHARMACEUTICAL SCIENCES Available online at: http://www.iajps.com Research Article DEVELOPMENT OF UV SPECTROPHOTOMETRIC METHOD FOR ESTIMATION OF SERATRODAST IN BULK AND PHARMACEUTICAL FORMULATIONS Naveen Kumar G S*, Harish K H, Hanumanthachar Joshi Sarada Vilas College of Pharmacy, K. M. Puram, Mysuru, India. Abstract: A simple UV spectrophotometric method has been developed for the estimation of Seratrodast pure form and pharmaceutical formulation. Seratrodast in bulk drug and pharmaceutical formulation and has an absorption maximum at 285nm in methanol. It obeys Beer’s law in the concentration range of 20 -100 µg/ml. The method was measured at its appropriate λmax against the reagent blank. The developed method was found to be precise, accurate and reproducible. Keywords: Beer’s law, Seratrodast, methanol and UV spectrophotometry *Corresponding author: Naveen Kumar G.S, QR code Sarada Vilas College of Pharmacy, K. M. Puram, Mysuru, India Email: [email protected], Please cite this article in press as Naveen Kumar G.S et al., Development of UV Spectrophotometric Method for Estimation of Seratrodastin Bulk and Pharmaceutical Formulations, Indo Am. J. P. Sci, 2018; 05(01). www.iajps.com Page 33 IAJPS 2018, 05 (01), 33-36 Naveen Kumar G.S et al ISSN 2349-7750 INTRODUCTION: Experimental methods Seratrodast is a thromboxane A2 (TXA2) receptor Preparation of Standard stock solution (TP receptor) antagonist used primarily in the Standard solution of seratrodast (1 mg/ml) was treatment of asthma. -

Mechanisms of Cyclooxygenase-2-Dependent Human Aortic Smooth Muscle Cell Phenotypic Modulation
University of Kentucky UKnowledge Theses and Dissertations--Pharmacy College of Pharmacy 2014 MECHANISMS OF CYCLOOXYGENASE-2-DEPENDENT HUMAN AORTIC SMOOTH MUSCLE CELL PHENOTYPIC MODULATION Oreoluwa O. Adedoyin University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Adedoyin, Oreoluwa O., "MECHANISMS OF CYCLOOXYGENASE-2-DEPENDENT HUMAN AORTIC SMOOTH MUSCLE CELL PHENOTYPIC MODULATION" (2014). Theses and Dissertations--Pharmacy. 34. https://uknowledge.uky.edu/pharmacy_etds/34 This Doctoral Dissertation is brought to you for free and open access by the College of Pharmacy at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Pharmacy by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. I agree that the document mentioned above may be made available immediately for worldwide access unless an embargo applies. -

Different Prostanoids Are Involved in Bradykinin-Induced Endothelium-Dependent and -Independent Vasoconstriction in Rat Mesenteric Resistance Arteries
J Pharmacol Sci 94, 115 – 121 (2004) Journal of Pharmacological Sciences ©2004 The Japanese Pharmacological Society Full Paper Different Prostanoids Are Involved in Bradykinin-Induced Endothelium-Dependent and -Independent Vasoconstriction in Rat Mesenteric Resistance Arteries Hideki Nawa1, Yuji Kurosaki1, and Hiromu Kawasaki1,* 1Department of Clinical Pharmaceutical Science, Graduate School of Natural Science and Technology, Okayama University, 1-1-1 Tsushima-naka, Okayama 700-8530, Japan Received December 3, 2002; Accepted December 3, 2003 Abstract. Mechanisms underlying bradykinin-induced vasoconstriction were investigated in rat perfused mesenteric vascular beds with active tone. In preparations with intact endothelium, bolus injections of bradykinin (1 to 1,000 pmol) dose-dependently produced three-phase vascular effects, which consisted of a first-phase vasodilation followed by a second-phase vasoconstric- tion and a subsequent third-phase vasodilation; these effects were abolished by FR172357 9 8 (bradykinin B2-receptor antagonist), but not by des-Arg -[Leu ]-bradykinin (bradykinin B1-recep- tor antagonist). In preparations with intact endothelium, indomethacin (cyclooxygenase inhibi- tor), seratrodast (thromboxane A2 (TXA2)-receptor antagonist), ONO-3708 (TXA2/prostaglandin H2 (PGH2)-receptor antagonist) or ozagrel (TXA2 synthesis inhibitor) markedly inhibited the bradykinin-induced vasoconstriction. In preparations without endothelium, the bradykinin-induced vasoconstriction was abolished by indomethacin and ONO-3708, while seratrodast -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. -

(12) Patent Application Publication (10) Pub. No.: US 2004/0058896 A1 Dietrich Et Al
US 200400.58896A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2004/0058896 A1 Dietrich et al. (43) Pub. Date: Mar. 25, 2004 (54) PHARMACEUTICAL PREPARATION (30) Foreign Application Priority Data COMPRISING AN ACTIVE DISPERSED ON A MATRIX Dec. 7, 2000 (EP)........................................ OO126847.3 (76) Inventors: Rango Dietrich, Konstanz (DE); Publication Classification Rudolf Linder, Kontanz (DE); Hartmut Ney, Konstanz (DE) (51) Int. Cl." ...................... A61K 31156; A61K 31/4439 (52) U.S. Cl. ........................... 514/171; 514/179; 514/338 Correspondence Address: (57) ABSTRACT NATH & ASSOCATES PLLC 1030 FIFTEENTH STREET, N.W. The present invention relates to the field of pharmaceutical SIXTH FLOOR technology and describes a novel advantageous preparation WASHINGTON, DC 20005 (US) for an active ingredient. The novel preparation is Suitable for 9 producing a large number of pharmaceutical dosage forms. (21) Appl. No.: 10/433,398 In the new preparation an active ingredient is present essentially uniformly dispersed in an excipient matrix com (22) PCT Filed: Dec. 6, 2001 posed of one or more excipients Selected from the group of fatty alcohol, triglyceride, partial glyceride and fatty acid (86) PCT No.: PCT/EPO1/14307 eSter. US 2004/0058896 A1 Mar. 25, 2004 PHARMACEUTICAL PREPARATION 0008 Further subject matters are evident from the claims. COMPRISING AN ACTIVE DISPERSED ON A MATRIX 0009. The preparations for the purpose of the invention preferably comprise numerous individual units in which at least one active ingredient particle, preferably a large num TECHNICAL FIELD ber of active ingredient particles, is present in an excipient 0001. The present invention relates to the field of phar matrix composed of the excipients of the invention (also maceutical technology and describes a novel advantageous referred to as active ingredient units hereinafter). -

The Biology of Prostaglandins and Their Role As a Target for Allergic Airway Disease Therapy
International Journal of Molecular Sciences Review The Biology of Prostaglandins and Their Role as a Target for Allergic Airway Disease Therapy Kijeong Lee, Sang Hag Lee and Tae Hoon Kim * Department of Otorhinolaryngology-Head & Neck Surgery, College of Medicine, Korea University, Seoul 02841, Korea; [email protected] (K.L.); [email protected] (S.H.L.) * Correspondence: [email protected]; Tel.: +82-02-920-5486 Received: 23 January 2020; Accepted: 5 March 2020; Published: 8 March 2020 Abstract: Prostaglandins (PGs) are a family of lipid compounds that are derived from arachidonic acid via the cyclooxygenase pathway, and consist of PGD2, PGI2, PGE2, PGF2, and thromboxane B2. PGs signal through G-protein coupled receptors, and individual PGs affect allergic inflammation through different mechanisms according to the receptors with which they are associated. In this review article, we have focused on the metabolism of the cyclooxygenase pathway, and the distinct biological effect of each PG type on various cell types involved in allergic airway diseases, including asthma, allergic rhinitis, nasal polyposis, and aspirin-exacerbated respiratory disease. Keywords: prostaglandins; allergy; asthma; allergic rhinitis; AERD; PGD2; PGE2 1. Introduction Prostaglandins (PGs) are lipid mediators, generated from arachidonic acid (AA) metabolism via cyclooxygenases (COX). They were discovered in the 1930s as regulators of blood pressure and smooth muscle contraction [1]. The distribution of synthases and receptors for each PG is different in various cell types, and PGs are activated via either paracrine or autocrine signaling on the surface of each cell type [2]. PGs bridge the interactions between various immune-modulating cells, and are considered key players in regulating pro-inflammatory and anti-inflammatory responses [3]. -

(12) Patent Application Publication (10) Pub. No.: US 2014/0328917 A1 Tada Et Al
US 20140328917A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0328917 A1 Tada et al. (43) Pub. Date: Nov. 6, 2014 (54) ORGANIC COMPOUND NANO-POWDER, BO2C23/8 (2006.01) METHOD FOR PRODUCING THE SAME AND A63/67 (2006.01) SUSPENSION A63L/92 (2006.01) A613 L/405 (2006.01) (71) Applicant: Activus Pharma Co., Ltd., A613 L/4I (2006.01) Funabashi-shi, Chiba (JP) A 6LX3/573 (2006.01) A613 L/7048 (2006.01) (72) Inventors: Takahiro Tada, Funabashi-shi (JP); A63L/96 (2006.01) Kazuhiro Kagami, Funabashi-shi (JP): (52) U.S. Cl. Shiro Yokota, Funabashi-shi (JP) CPC ........... A61 K9/1682 (2013.01); A61 K3I/7048 (2013.01); A61 K3I/445 (2013.01); A61 K (21) Appl. No.: 14/001,645 3 1/57 (2013.01); A61 K3I/I2 (2013.01); A61 K 31/196 (2013.01); A61K 31/167 (2013.01); (22) PCT Filed: May 11, 2013 A6 IK3I/192 (2013.01); A61 K3I/405 (86). PCT No.: PCT/UP2013/003023 (2013.01); A61 K3I/41 (2013.01); A61 K 31/573 (2013.01); B02C 23/18 (2013.01) S371 (c)(1), USPC 424/489: 514/27: 514/317; 514/179; 514/29: (2), (4) Date: Aug. 26, 2013 514/679:514/567; 514/629; 514/570; 514/31; 514/.420, 514/382: 514/180; 24.1/15 (30) Foreign Application Priority Data (57) ABSTRACT May 11, 2012 (JP) ................................. 2012-108.972 An organic compound nano-powder comprising a granular organic compound with an average particle diameter of 500 Publication Classification nm or less and a 90%-diameter of less than 1500 nm and a carbohydrate compound comprising at least any one of a (51) Int. -

(12) United States Patent (10) Patent No.: US 8,506,935 B2 Hale Et Al
US008506935B2 (12) United States Patent (10) Patent No.: US 8,506,935 B2 Hale et al. (45) Date of Patent: *Aug. 13, 2013 (54) RESPIRATORY DRUG CONDENSATION 4,848,374. A 7, 1989 Chard et al. AEROSOLS AND METHODS OF MAKING 16 A 3. R; A. tal AND USING THEMI 4,906,417w- - A 3/1990 Gentry SU ca. 4,917,119 A 4, 1990 Potter et al. (75) Inventors: Ron L. Hale, Sandia Park, NM (US); 4,924,883. A 5/1990 Perfetti et al. Peter M. Lloyd, Walnut Creek, CA 4.941,483. A 7/1990 Ridings et al. (US); Amy T. Lu, Los Altos, CA (US); 3. A g 3. R. et t 1 Joshua D. Rabinowitz, Princeton, NJ 5,049,389W 4 A 9, 1991 Radhakrishnunanerjee et al. (US); Martin J. Wensley, Los Gatos, 5,060,671. A 10/1991 Counts et al. CA (US) 5,099,861. A 3/1992 Clearman et al. 5,135,009 A 8, 1992 Muller et al. (73) Assignee: Alexza Pharmaceuticals, Inc., : A 2. 3: R et al. Mountain rView, ar. CA (US) 5,190,029J. W. A 3/1993 Byronetontgomery al. 5,224,498 A 7/1993 Deevi et al. (*) Notice: Subject to any disclaimer, the term of this 5,345,951 A 9, 1994 G al. patent is extended or adjusted under 35 5,366,770 A 1 1/1994 Wang U.S.C. 154(b) by 134 days. 5,388,574 A 2/1995 Ingebrethsen 5,456,247 A 10/1995 Shilling et al. This patent is Subject to a terminal dis- 5,511,726 A 4/1996 Greenspan et al. -
Chemical Structure-Related Drug-Like Criteria of Global Approved Drugs
Molecules 2016, 21, 75; doi:10.3390/molecules21010075 S1 of S110 Supplementary Materials: Chemical Structure-Related Drug-Like Criteria of Global Approved Drugs Fei Mao 1, Wei Ni 1, Xiang Xu 1, Hui Wang 1, Jing Wang 1, Min Ji 1 and Jian Li * Table S1. Common names, indications, CAS Registry Numbers and molecular formulas of 6891 approved drugs. Common Name Indication CAS Number Oral Molecular Formula Abacavir Antiviral 136470-78-5 Y C14H18N6O Abafungin Antifungal 129639-79-8 C21H22N4OS Abamectin Component B1a Anthelminithic 65195-55-3 C48H72O14 Abamectin Component B1b Anthelminithic 65195-56-4 C47H70O14 Abanoquil Adrenergic 90402-40-7 C22H25N3O4 Abaperidone Antipsychotic 183849-43-6 C25H25FN2O5 Abecarnil Anxiolytic 111841-85-1 Y C24H24N2O4 Abiraterone Antineoplastic 154229-19-3 Y C24H31NO Abitesartan Antihypertensive 137882-98-5 C26H31N5O3 Ablukast Bronchodilator 96566-25-5 C28H34O8 Abunidazole Antifungal 91017-58-2 C15H19N3O4 Acadesine Cardiotonic 2627-69-2 Y C9H14N4O5 Acamprosate Alcohol Deterrant 77337-76-9 Y C5H11NO4S Acaprazine Nootropic 55485-20-6 Y C15H21Cl2N3O Acarbose Antidiabetic 56180-94-0 Y C25H43NO18 Acebrochol Steroid 514-50-1 C29H48Br2O2 Acebutolol Antihypertensive 37517-30-9 Y C18H28N2O4 Acecainide Antiarrhythmic 32795-44-1 Y C15H23N3O2 Acecarbromal Sedative 77-66-7 Y C9H15BrN2O3 Aceclidine Cholinergic 827-61-2 C9H15NO2 Aceclofenac Antiinflammatory 89796-99-6 Y C16H13Cl2NO4 Acedapsone Antibiotic 77-46-3 C16H16N2O4S Acediasulfone Sodium Antibiotic 80-03-5 C14H14N2O4S Acedoben Nootropic 556-08-1 C9H9NO3 Acefluranol Steroid