UTR Interactions Regulate P53 Mrna Translation and Provide a Target for Modulating P53 Induction After DNA Damage
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Susceptibility of Heterozygous and Nullizygous P53 Knockout Mice to Chemical Carcinogens: Tissue Dependence and Role of P53 Gene Mutations
J Toxicol Pathol 2005; 18: 121–134 Review Susceptibility of Heterozygous and Nullizygous p53 Knockout Mice to Chemical Carcinogens: Tissue Dependence and Role of p53 Gene Mutations Tetsuya Tsukamoto1, Akihiro Hirata1, and Masae Tatematsu1 1Division of Oncological Pathology, Aichi Cancer Center Research Institute, 1–1 Kanokoden, Chikusa-ku, Nagoya 464–8681, Japan Abstract: Mutations of the p53 tumor suppressor gene constitute one of the most frequent molecular changes in a wide variety of human cancers and mice deficient in p53 have recently attracted attention for their potential to identify chemical genotoxins. In this article, we review the data on the susceptibility of p53 nullizygote (–/–), heterozygote (+/ –), and wild type (+/+) mice to various carcinogens. Induction of esophageal and tongue squamous cell carcinomas (SCCs) by methyl-n-amylnitrosamine was shown to be increased in p53 (+/–) mice, in addition to the high sensitivity shown by p53 (–/–) littermates. N,N-dibutylnitrosamine (DBN) treatment also caused more tumor development in p53 (+/–) than wild-type mice, as with N-butyl-N-(4-hydroxybutyl)nitrosamine (BBN) in the urinary bladder. In addition, p53 (+/–) heterozygotes proved more sensitive than wild type littermates to the induction of stromal cell tumors like hemangiomas/hemangiosarcomas by N-bis(2-hydroxypropyl)nitrosamine (BHP) or lymphomas and fibrosarcomas with other carcinogens. Analysis of exons 5–8 of the p53 gene demonstrated mutations in approximately one half of the lesions. With N-methyl-N-nitrosourea, preneoplastic lesions of the stomach, pepsinogen altered pyloric glands (PAPG), and a gastric adenocarcinoma, were found after only 15 weeks in p53 (–/–) mice, although there was no significant difference in the incidence of gastric tumors between p53 (+/+) and (+/–) mice in the longer-term. -

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

Gene Expression Patterns Define Novel Roles for E47 in Cell Cycle Progression, Cytokine-Mediated Signaling, and T Lineage Development
Gene expression patterns define novel roles for E47 in cell cycle progression, cytokine-mediated signaling, and T lineage development Ruth Schwartz*†, Isaac Engel*†‡, Mohammad Fallahi-Sichani§, Howard T. Petrie§, and Cornelis Murre*¶ *Division of Biological Sciences, University of California at San Diego, La Jolla, CA 92093-0377; and §Scripps͞Florida Research Institute, 5353 Parkside Drive, RF-1, Jupiter, FL 33458 Communicated by Michael S. Levine, University of California, Berkeley, CA, May 5, 2006 (received for review April 7, 2006) In maturing T lineage cells, the helix–loop–helix protein E47 has analyze the global patterns of gene expression in E2A-deficient been shown to enforce a critical proliferation and developmental lymphomas in the absence and presence of E47. Cluster analysis checkpoint commonly referred to as  selection. To examine how identified groups of genes that were regulated by E47 and shared E47 regulates cellular expansion and developmental progression, biochemical or physiological properties. These included genes we have used an E2A-deficient lymphoma cell line and DNA involved in cell survival, cell cycle progression, stress response, lipid microarray analysis to identify immediate E47 target genes. Hier- metabolism, ␣, and natural killer T (NKT) cell function. Based on archical cluster analysis of gene expression patterns revealed that these observations, we suggest that E47 functions in molecular E47 coordinately regulates the expression of genes involved in cell pathways to coordinate cell survival, cell growth, and developmen- survival, cell cycle progression, lipid metabolism, stress response, tal maturation. We identified cyclin-dependent kinase 6 (Cdk6) as and lymphoid maturation. These include Plc␥2, Cdk6, CD25, Tox, an E47 target gene, providing a potential mechanism underlying a Gadd45a, Gadd45b, Gfi1, Gfi1b, Socs1, Socs3, Id2, Eto2, and Xbp1. -

Supplementary Material Contents
Supplementary Material Contents Immune modulating proteins identified from exosomal samples.....................................................................2 Figure S1: Overlap between exosomal and soluble proteomes.................................................................................... 4 Bacterial strains:..............................................................................................................................................4 Figure S2: Variability between subjects of effects of exosomes on BL21-lux growth.................................................... 5 Figure S3: Early effects of exosomes on growth of BL21 E. coli .................................................................................... 5 Figure S4: Exosomal Lysis............................................................................................................................................ 6 Figure S5: Effect of pH on exosomal action.................................................................................................................. 7 Figure S6: Effect of exosomes on growth of UPEC (pH = 6.5) suspended in exosome-depleted urine supernatant ....... 8 Effective exosomal concentration....................................................................................................................8 Figure S7: Sample constitution for luminometry experiments..................................................................................... 8 Figure S8: Determining effective concentration ......................................................................................................... -

Delayed Treatment with 4-Methylpyrazole Protects Against Acetaminophen Hepatotoxicity in Mice by Inhibition of C-Jun N-Terminal Kinase Jephte Y
TOXICOLOGICAL SCIENCES, 170(1), 2019, 57–68 doi: 10.1093/toxsci/kfz077 Advance Access Publication Date: March 23, 2019 Research Article Delayed Treatment With 4-Methylpyrazole Protects Against Acetaminophen Hepatotoxicity in Mice by Inhibition of c-Jun n-Terminal Kinase Jephte Y. Akakpo,* Anup Ramachandran,* Luqi Duan,* Matthew A. Schaich,† Matthew W. Jaeschke,* Bret D. Freudenthal,† Wen-Xing Ding,* Barry H. Rumack,‡ and Hartmut Jaeschke*,1 *Department of Pharmacology Toxicology & Therapeutics and †Department of Biochemistry and Molecular Biology, University of Kansas Medical Center, Kansas City, Kansas 66160; and ‡Department of Emergency Medicine and Pediatrics, University of Colorado School of Medicine, Aurora, Colorado 80045 1To whom correspondence should be addressed at Department of Pharmacology, Toxicology & Therapeutics, University of Kansas Medical Center, 3901 Rainbow Blvd, MS 1018, Kansas City, KS 66160. Fax 913 588 7501; E-mail: [email protected]. ABSTRACT Acetaminophen (APAP) overdose is the most common cause of hepatotoxicity and acute liver failure in the United States and many western countries. However, the only clinically approved antidote, N-acetylcysteine, has a limited therapeutic window. 4-Methylpyrazole (4MP) is an antidote for methanol and ethylene glycol poisoning, and we have recently shown that cotreatment of 4MP with APAP effectively prevents toxicity by inhibiting Cyp2E1. To evaluate if 4MP can be used therapeutically, C57BL/6J mice were treated with 300 mg/kg APAP followed by 50 mg/kg 4MP 90 min later (after the metabolism phase). In these experiments, 4MP significantly attenuated liver injury at 3, 6, and 24 h after APAP as shown by 80%–90% reduction in plasma alanine aminotransferase activities and reduced areas of necrosis. -

Immune Suppressive Landscape in the Human Esophageal Squamous Cell
ARTICLE https://doi.org/10.1038/s41467-020-20019-0 OPEN Immune suppressive landscape in the human esophageal squamous cell carcinoma microenvironment ✉ Yingxia Zheng 1,2,10 , Zheyi Chen1,10, Yichao Han 3,10, Li Han1,10, Xin Zou4, Bingqian Zhou1, Rui Hu5, Jie Hao4, Shihao Bai4, Haibo Xiao5, Wei Vivian Li6, Alex Bueker7, Yanhui Ma1, Guohua Xie1, Junyao Yang1, ✉ ✉ ✉ Shiyu Chen1, Hecheng Li 3 , Jian Cao 7,8 & Lisong Shen 1,9 1234567890():,; Cancer immunotherapy has revolutionized cancer treatment, and it relies heavily on the comprehensive understanding of the immune landscape of the tumor microenvironment (TME). Here, we obtain a detailed immune cell atlas of esophageal squamous cell carcinoma (ESCC) at single-cell resolution. Exhausted T and NK cells, regulatory T cells (Tregs), alternatively activated macrophages and tolerogenic dendritic cells are dominant in the TME. Transcriptional profiling coupled with T cell receptor (TCR) sequencing reveal lineage con- nections in T cell populations. CD8 T cells show continuous progression from pre-exhausted to exhausted T cells. While exhausted CD4, CD8 T and NK cells are major proliferative cell components in the TME, the crosstalk between macrophages and Tregs contributes to potential immunosuppression in the TME. Our results indicate several immunosuppressive mechanisms that may be simultaneously responsible for the failure of immuno-surveillance. Specific targeting of these immunosuppressive pathways may reactivate anti-tumor immune responses in ESCC. 1 Department of Laboratory Medicine, Xin Hua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China. 2 Institute of Biliary Tract Diseases Research, Shanghai Jiao Tong University School of Medicine, Shanghai, China. 3 Department of Thoracic Surgery, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China. -

Pharmacology Review(S) Memorandum
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 207103Orig1s000 PHARMACOLOGY REVIEW(S) MEMORANDUM Ibrance (palbociclib) Date: January 23, 2015 To: File for NDA 207103 From: John K. Leighton, PhD, DABT Acting Director, Division of Hematology Oncology Toxicology Office of Hematology and Oncology Products I have examined pharmacology/toxicology supporting and labeling reviews for Ibrance conducted by Dr. Chen, and secondary memorandum and labeling provided by Dr. Palmby. I concur with Dr. Palmby’s conclusion that Ibrance may be approved and that no additional nonclinical studies are needed for the proposed indication. Reference ID: 3691405 --------------------------------------------------------------------------------------------------------- This is a representation of an electronic record that was signed electronically and this page is the manifestation of the electronic signature. --------------------------------------------------------------------------------------------------------- /s/ ---------------------------------------------------- JOHN K LEIGHTON 01/23/2015 Reference ID: 3691405 MEMORANDUM Date: January 22, 2015 From: Todd R. Palmby, PhD Pharmacology/Toxicology Supervisor Division of Hematology Oncology Toxicology (DHOT) Office of Hematology and Oncology Products (OHOP) To: File for NDA 207103 Ibrance (palbociclib) Re: Approvability for Pharmacology and Toxicology Indication: in combination with letrozole for the treatment of postmenopausal women with estrogen receptor (ER)-positive, human epidermal growth factor -

Review Recognition of Specific DNA Sequences
Molecular Cell, Vol. 8, 937–946, November, 2001, Copyright 2001 by Cell Press Recognition of Specific DNA Sequences Review Colin W. Garvie and Cynthia Wolberger1 teins that mediate recombination, DNA cleavage, meth- Department of Biophysics and Biophysical Chemistry ylation, and other processes. The broad principles of and the Howard Hughes Medical Institute DNA binding outlined here pertain to these proteins as Johns Hopkins University School of Medicine well, although the structural details differ. Baltimore, Maryland 21205 Basic Requirements for DNA Binding Double-stranded DNA is a polymer of relatively uniform Proteins that recognize specific DNA sequences play structure, with a highly negatively charged sugar-phos- a central role in the regulation of transcription. The phate backbone and a core of stacked base pairs whose tremendous increase in structural information on pro- edges are exposed in the major and minor grooves. tein-DNA complexes has uncovered a remarkable Since each base pair has a characteristic set of func- structural diversity in DNA binding folds, while at the tional groups, each DNA sequence has a chemical “sig- same time revealing common themes in binding to nature” characterized by the pattern of these groups target sites in the genome. exposed in the DNA grooves. It is this chemical surface, along with sequence-dependent variations in DNA Introduction structure and flexibility, that is recognized by proteins. Proteins recognize a particular sequence by having a The ability of a protein to bind selectively to a particular surface that is chemically complementary to that of the DNA site in the genome is the foundation upon which DNA, forming a series of favorable electrostatic and van transcriptional regulatory pathways are built. -

Early Life Exposure to Ethinylestradiol Enhances Subsequent Responses To
www.nature.com/scientificreports OPEN Early life exposure to ethinylestradiol enhances subsequent responses to Received: 18 August 2017 Accepted: 4 January 2018 environmental estrogens measured Published: xx xx xxxx in a novel transgenic zebrafsh Jon M. Green1, Anke Lange 1, Aaron Scott1, Maciej Trznadel1, Htoo Aung Wai 1, Aya Takesono1, A. Ross Brown 1, Stewart F. Owen2, Tetsuhiro Kudoh1 & Charles R. Tyler1 Estrogen plays fundamental roles in a range of developmental processes and exposure to estrogen mimicking chemicals has been associated with various adverse health efects in both wildlife and human populations. Estrogenic chemicals are found commonly as mixtures in the environment and can have additive efects, however risk analysis is typically conducted for single-chemicals with little, or no, consideration given for an animal’s exposure history. Here we developed a transgenic zebrafsh with a photoconvertable fuorophore (Kaede, green to red on UV light exposure) in a skin pigment-free mutant element (ERE)-Kaede-Casper model and applied it to quantify tissue-specifc fuorescence biosensor responses for combinations of estrogen exposures during early life using fuorescence microscopy and image analysis. We identify windows of tissue-specifc sensitivity to ethinylestradiol (EE2) for exposure during early-life (0–5 dpf) and illustrate that exposure to estrogen (EE2) during 0–48 hpf enhances responsiveness (sensitivity) to diferent environmental estrogens (EE2, genistein and bisphenol A) for subsequent exposures during development. Our fndings illustrate the importance of an organism’s stage of development and estrogen exposure history for assessments on, and possible health risks associated with, estrogen exposure. Exposure to endocrine disrupting chemicals (EDCs) is linked with a range of adverse health disorders and further understanding of EDCs efects is crucial for safe-guarding long-term human and environmental health1,2. -
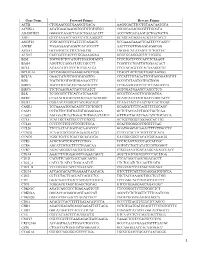
Gene Name Forward Primer Reverse Primer ACTB
Gene Name Forward Primer Reverse Primer ACTB CTGGAACGGTGAAGGTGACA AAGGGACTTCCTGTAACAATGCA ACVRL1 ACATGAAGAAGGTGGTGTGTGTGG CGGGCAGAGGGGTTTGGGTA ADAMDEC1 GGGGCCAGACTACACTGAAACATT ACCCGTCACAAGTACTGATGCTG AHI1 GTCCAAAACTACCCCATCAAGGCT GCAGCACAGGAACGTATCACCT ANGPT2 TGGCAGCGTTGATTTTCAGAGG GCGAAACAAACTCATTTCCCAGCC ANPEP TGAAGAAGCAGGTCACACCCCT AACTCCGTTGGAGCAGGCGG APOA1 GCCGTGCTCTTCCTGACGG TGGGACACATAGTCTCTGCCGC ATXN7 CACCGCCCACTCTGGAAAAGAA GGGTGCAGGGCTTCTTGGTG B2M TGCTGTCTCCATGTTTGATGTATCT TCTCTGCTCCCCACCTCTAAGT BAG4 AGGTTCCAGGATATCCGCCTT TCGGTCCTGATTGTGGAACACT BCL2 ACAACATCGCCCTGTGGATGA CCGTACAGTTCCACAAAGGCAT BCL2L14 GCTCAGGGTCAAAGGACGTTGG TCAGCTACTCGGTTGGCAATGG BCL7A GAACCATGTCGGGCAGGTCG CCCATTTGTAGATTCGTAGGGATGTGT BIN1 TGCTGTCGTGGTGGAGACCTTC GCCGTGTAGTCGTGCTGGG BIRC3 TGCTATCCACATCAGACAGCCC TCTGAATGGTCTTCTCCAGGTTCA BIRC5 TTCTCAAGGACCACCGCATCT AGTGGATGAAGCCAGCCTCG BLK TCGGGGTCTTCACCATCAAAGC GCGCTCCAGGTTGCGGATGA BTRC CCAAATGTGTCATTACCAACATGGGC GCAGCACATAGTGATTTGGCATCC BUB3 CGGAACATGGGTTACGTGCAGC CCAAATACTCAACTGCCACTCGGC CAGE1 TCCAAAATGCACAGTCTTCTGGCT GGAGGCTCTTCAGTTTTTGCAGC CASP1 CCTGTTCCTGTGATGTGGAGGAAA GCTCTACCATCTGGCTGCTCAA CASP3 AGCGAATCAATGGACTCTGGAATATCC GTTTGCTGCATCGACATCTGTACCA CCL5 TCATTGCTACTGCCCTCTGCG ACTGCTGGGTTGGAGCACTTG CCL18 CCCTCCTTGTCCTCGTCTGCA GCACTGGGGGCTGGTTTCAG CCL26 TTCCAATACAGCCACAAGCCCC GGATGGGTACAGACTTTCTTGCCTC CCND2 TCAAGTGCGTGCAGAAGGACAT CTTCGCACTTCTGTTCCTCACA CCND3 TGGCTGCTGTGATTGCACATGA GATGGCGGGTACATGGCAAAGG CCR3 ACGCTGCTCTGCTTCCTGG TCCTCAGTTCCCCACCATCGC CCR4 AGCATCGTGCTTCCTGAGCAA GGTGTCTGCTATATCCGTGGGGT CCR7 AGACAGGGGTAGTGCGAGGC -

Eomes Transcription Factor Is Required for the Development and Differentiation of Invariant NKT Cells
ARTICLE https://doi.org/10.1038/s42003-019-0389-3 OPEN Eomes transcription factor is required for the development and differentiation of invariant NKT cells Kanako Shimizu1,3, Yusuke Sato1,3, Masami Kawamura1, Hiroshi Nakazato1, Takashi Watanabe2, 1234567890():,; Osamu Ohara2 & Shin-ichiro Fujii1,2 Eomes regulates the differentiation of CD8+ T cells into effector and memory phases. However, its role in invariant (i)NKT cells remains unknown. Here, we show the impact of Eomes on iNKT cells in the thymus and peripheral tissue using conditional knockout (Eomes- cKO) mice. In the thymus, CD1d-tetramer+CD24+CD44−NK1.1−CD69+stage 0 iNKT cells express higher levels of Eomes than the other iNKT stages. We also found that Eomes regulates NKT1 cell differentiation predominantly. Interestingly, the expression of Eomes in the steady state is low, but can be upregulated after TCR stimulation. We also showed epigenetic changes in the Eomes locus after activation. In addition, vaccination of C57BL/6, but not Eomes-cKO mice with iNKT ligand-loaded dendritic cells generated KLRG1 +iNKT cells in lung, characterized as effector memory phenotype by transcriptome profiling. Thus, Eomes regulates not only the differentiation of NKT1 cells in the thymus, but also their differentiation into memory-like KLRG1+iNKT cells in the periphery. 1 Laboratory for Immunotherapy, RIKEN Center for Integrative Medical Sciences (IMS), Yokohama, Kanagawa 230-0045, Japan. 2 Laboratory for Integrative Genomics, RIKEN Center for Integrative Medical Sciences (IMS), Yokohama, Kanagawa 230-0045, Japan. 3These authors contributed equally: Kanako Shimizu, Yusuke Sato. Correspondence and requests for materials should be addressed to K.S. -

Counteracting CAR T Cell Dysfunction
Oncogene (2021) 40:421–435 https://doi.org/10.1038/s41388-020-01501-x REVIEW ARTICLE Counteracting CAR T cell dysfunction 1,2 1 1 1 2 Mansour Poorebrahim ● Jeroen Melief ● Yago Pico de Coaña ● Stina L. Wickström ● Angel Cid-Arregui ● Rolf Kiessling 1 Received: 6 May 2020 / Revised: 22 September 2020 / Accepted: 30 September 2020 © The Author(s) 2020. This article is published with open access Abstract In spite of high rates of complete remission following chimeric antigen receptor (CAR) T cell therapy, the efficacy of this approach is limited by generation of dysfunctional CAR T cells in vivo, conceivably induced by immunosuppressive tumor microenvironment (TME) and excessive antigen exposure. Exhaustion and senescence are two critical dysfunctional states that impose a pivotal hurdle for successful CAR T cell therapies. Recently, modified CAR T cells with an “exhaustion- resistant” phenotype have shown superior antitumor functions and prolonged lifespan. In addition, several studies have indicated the feasibility of senescence delay in CAR T cells. Here, we review the latest reports regarding blockade of CAR T cell exhaustion and senescence with a particular focus on the exhaustion-inducing pathways. Subsequently, we describe what potential these latest insights offer for boosting the potency of adoptive cell transfer (ACT) therapies involving CAR T cells. 1234567890();,: 1234567890();,: Furthermore, we discuss how induction of costimulation, cytokine exposure, and TME modulation can impact on CAR T cell efficacy and persistence, while potential safety issues associated with reinvigorated CAR T cells will also be addressed. Introduction persistence in the hostile tumor microenvironment (TME) represent additional key impediments to efficacious CAR T In a recent decade, genetically modified immune cells, parti- cell responses and durable clinical remission following CAR T cularly chimeric antigen receptor (CAR) T cells, have raised cell therapy [4].