Influenza Virus-Specific CD8+ T Cells -Longevity, Cross-Reactivity and Viral Evasion
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
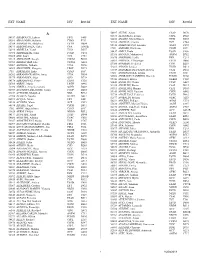
Ext Name Div Room Ext Name Div Room ______
EXT NAME DIV ROOM EXT NAME DIV ROOM __________________________________________________________________________________________________________________________ A 56407 ALUNG, Ashok CSAP D170 52673 ALVETRETI, Letizia CSPL D328 54157 ABABOUCH, Lahsen FIPX F405 54458 AMADO, Maria Blanca TCIB D503 55581 ABALSAMO, Stefania CIOO F713 53290 AMARAL, Cristina TCE C744 53264 ABBASSIAN, Abdolreza ESTD D804 55126 AMBROSIANO, Luciana AGDF C292 54511 ABBONDANZA, Carla CPA A143B 1709 AMEGEE, Emilienne CSSD SSC 52390 ABDELLA, Yesuf TCIA D557 56437 AMICI, Paolo ESSD C456 55396 ABDIRIZZAK, Tania FOED C474 53985 AMMATI, Mohammed AGPM B752 53485 ABE, Kaori TCE C751 54876 AMOROSO, Leslie ESN C214 54315 ABI NASSIF, Joseph CSDM B104 56891 AMROUK, El Mamoun ESTD D866 53963 ABI RACHED, Elie CPAM A281 53980 ANAMAN, Frederick CIOF B239 55087 ABITBOL, Nathalie TCIA D556 54213 ANAND, Sanjeev TCSD D611 56584 ABOU-RIZK, Margaret LEGP D371 53309 ANDARIAS DE PRADO, Rebeca CSAI D102 1716 ABRAHAM, Erika CSSD SSC 1725 ANDONOVSKA, Liljana CSSD SSC 56202 ABRAMO-GUARNA, Anna TCIA D554 53602 ANDRADE CIANFRINI, Graciela FOMD D468 55375 ABRAMOVA, Olga OSD D728 55742 ANELLO, Enrico OEKM C102 54178 ABRAMOVICI, Pierre CIOH C130 54441 ANGELINI, Chiara CPAP A487 56760 ABREU, Maria LEGD A442 54109 ANGELINI, Flavio CSAI D114 53296 ABRINA, Angelica Gavina OSPD B449 55313 ANGELONI, Marino CSAI D103 52709 ACCARDO-DELHOME, Jeanne FODP D435 53145 ANGELOZZI, Vanessa CSDU A042 53843 ACHOURI, Moujahed NRL B732 53720 ANGELUCCI, Federica ESTD D863 52294 ACOSTA, Natalia OEDD B452 52677 ANIBALDI, -

6 Chronic Abdominal Pain in Children
6 Chronic Abdominal Pain in Children Chronic Abdominal Pain in Children in Children Pain Abdominal Chronic Chronische buikpijn bij kinderen Carolien Gijsbers Carolien Gijsbers Carolien Chronic Abdominal Pain in Children Chronische buikpijn bij kinderen Carolien Gijsbers Promotiereeks HagaZiekenhuis Het HagaZiekenhuis van Den Haag is trots op medewerkers die fundamentele bijdragen leveren aan de wetenschap en stimuleert hen daartoe. Om die reden biedt het HagaZiekenhuis promovendi de mogelijkheid hun dissertatie te publiceren in een speciale Haga uitgave, die onderdeel is van de promotiereeks van het HagaZiekenhuis. Daarnaast kunnen promovendi in het wetenschapsmagazine HagaScoop van het ziekenhuis aan het woord komen over hun promotieonderzoek. Chronic Abdominal Pain in Children Chronische buikpijn bij kinderen © Carolien Gijsbers 2012 Den Haag ISBN: 978-90-9027270-2 Vormgeving en opmaak De VormCompagnie, Houten Druk DR&DV Media Services, Amsterdam Printing and distribution of this thesis is supported by HagaZiekenhuis. All rights reserved. Subject to the exceptions provided for by law, no part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form by any means, electronic, mechanical, photocopying, recording or otherwise, without the written consent of the author. Chronic Abdominal Pain in Children Chronische buikpijn bij kinderen Carolien Gijsbers Proefschrift ter verkrijging van de graad van doctor aan de Erasmus Universiteit Rotterdam op gezag van de rector magnificus Prof.dr. H.G. Schmidt en volgens besluit van het College voor Promoties. De openbare verdediging zal plaatsvinden op donderdag 20 december 2012 om 15.30 uur door Carolina Francesca Maria Gijsbers geboren te Zierikzee Promotiecommisie Promotor: Prof.dr. H.A. -
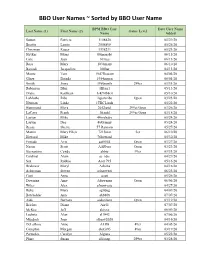
BBO User Names ~ Sorted by BBO User Name
BBO User Names ~ Sorted by BBO User Name BPM BBO User Date User Name Last Name (1) First Name (2) Game Level Name Added Sutton Patricia 1108820 05/29/20 Beattie Laurie 2006898 05/26/20 Chrisman Roger 3338231 05/21/20 Stryker Mona 00monabs 06/15/20 Cole Jean 101rye 06/13/20 Ross Mary 1038mary 06/10/20 Barsodi Jacqueline 168bar 04/11/20 Moore Tom 1947Deacon 04/04/20 Glaze Brenda 1954queen 06/01/20 Smith Jinny 1986north 299er 03/31/20 Robinson Blair 1Blair1 05/13/20 Drake Kathleen 1sK90b8c0 05/10/20 LaMothe Ribs 1spareribs Open 03/23/20 Herman Linda 1TBCLinda 05/22/20 Hammond Myra 2012ariel 299er/Open 03/26/20 LaCava Frank 3frankl 299er/Open 03/18/20 Layton Mike 44mybaby 05/24/20 Layton Dee 46Granny 05/24/20 Reese Sherre 57 Ransom 05/27/20 Martin Mary Ellen 7215mar 5er 06/10/20 Howard Mike 76howard 05/30/20 Fortado Avis aaf8844 Open 03/27/20 Nason Scott AAFlyer Open 03/23/20 Sterrantino Cyndy abbiy 99er 03/31/20 Cardinal Alain ac_idic 04/29/20 Sax Robbie Ace1795 05/16/20 Bralower Meryl Acksba 04/16/20 Ackerman Steven acksteveni 06/23/20 Curti Anne acurt 05/26/20 Downing Amy Adowning Open 06/06/20 Wiles Alex afwprivate 04/27/20 Ruhe Mary agildog 04/07/20 Batchelder Ann ahb808 07/07/20 Aide Barbara aidesifuen Open 03/19/20 Bricker Diane Air51 07/07/20 McKee Jeff akjxxx 06/09/20 Ludwig Alan al1942 07/06/20 Murdoch Joan albany2020 04/16/20 DeLaRosa Anne ALD8 49er 04/05/20 Compton Morgan alexis95 49er 03/19/20 Parmelee Carolyn Algona 05/22/20 Pharr Susan allisonp 299er 03/28/20 Hatch Deanna AlyceJake 04/21/20 Mason Annie amasonid 04/10/20 Flank -
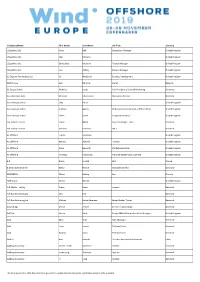
Company Name First Name Last Name Job Title Country
Company Name First Name Last Name Job Title Country 1StopWind Ltd Arran Bell Operations Manager United Kingdom 1StopWind Ltd. Alan Mckerns United Kingdom 1StopWind Ltd. Bernadette McAulay Finance Manager United Kingdom 1StopWind Ltd. Joel Telling General Manager United Kingdom 23 Degrees Renewables Ltd Ed Woodrow Business Development United Kingdom 24SEA bvba Gert De Sitter Owner Belgium 3S Europe GmbH Matthias Lamp Vice President of Sales & Marketing Germany 3sun Denmark ApS Christian Christensen Operations Director Denmark 3sun Group Limited Jody Potter United Kingdom 3sun Group Limited Graham Hacon VP Business Development, Offshore Wind United Kingdom 3sun Group Limited Sherri Smith Company Secretary United Kingdom 3W Industri Service Simon Øland Project manager - sales Denmark 3W Industri Service Kenneth Pedersen IWI-S Denmark 4C Offshore Lauren Anderson United Kingdom 4C Offshore Richard Aukland Director United Kingdom 4C Offshore Rosie Haworth Market Researcher United Kingdom 4C Offshore Vincenzo Poidomani Principal Geotechnical Engineer United Kingdom 8.2 Bruno ALLAIN CEO France 8.2 Monitoring GmbH Bernd Höring Managing director Germany 920338402 Ellinor Meling Ceo Norway A&P Group Emma Harrick United Kingdom A.P. Møller Holding Simon Ibsen Investor Denmark A/S Dan-Bunkering Ltd. Jens Kirk Denmark A/S Dan-Bunkering Ltd. Michael Brunø-Sørensen Senior Bunker Trader Denmark A1wind Aps Martin Jensen Director / A1wind Aps Denmark AAF Ltd Steven Brett Europe MFAS Aftermarket Sales Manager United Kingdom AAG Allan Tarp Sales Manager Denmark -

The Struggle of Three Generations Chinese Women for Claiming Identity Against Political Backdrop in Lian Gouw’S Only a Girl
PLAGIAT MERUPAKAN TINDAKAN TIDAK TERPUJI THE STRUGGLE OF THREE GENERATIONS CHINESE WOMEN FOR CLAIMING IDENTITY AGAINST POLITICAL BACKDROP IN LIAN GOUW’S ONLY A GIRL AN UNDERGRADUATE THESIS Presented as Partial Fulfillment of the Requirements for the Degree of Sarjana Sastra in English Letters By AJENG ANGGRAENI PUTRI Student Number: 134214002 ENGLISH LETTERS STUDY PROGRAM DEPARTMENT OF ENGLISH LETTERS FACULTY OF LETTERS SANATA DHARMA UNIVERSITY YOGYAKARTA 2017 PLAGIAT MERUPAKAN TINDAKAN TIDAK TERPUJI THE STRUGGLE OF THREE GENERATIONS CHINESE WOMEN FOR CLAIMING IDENTITY AGAINST POLITICAL BACKDROP IN LIAN GOUW’S ONLY A GIRL AN UNDERGRADUATE THESIS Presented as Partial Fulfillment of the Requirements for the Degree of Sarjana Sastra in English Letters By AJENG ANGGRAENI PUTRI Student Number: 134214002 ENGLISH LETTERS STUDY PROGRAM DEPARTMENT OF ENGLISH LETTERS FACULTY OF LETTERS SANATA DHARMA UNIVERSITY YOGYAKARTA 2017 ii PLAGIAT MERUPAKAN TINDAKAN TIDAK TERPUJI PLAGIAT MERUPAKAN TINDAKAN TIDAK TERPUJI PLAGIAT MERUPAKAN TINDAKAN TIDAK TERPUJI PLAGIAT MERUPAKAN TINDAKAN TIDAK TERPUJI PLAGIAT MERUPAKAN TINDAKAN TIDAK TERPUJI Difficult roads often lead to beautiful destinations~ “Two roads diverged in a wood, and I – I took the one less-traveled by, And that has made all the difference.” ~ Robert Frost “People are capable, at any time in their lives, of doing what they dream of.” ~ Paulo Coelho “Life ain’t always beautiful but it’s a beautiful ride.” ~ Gary Allan “I might only have one match but I can make an explosion.” ~ Rachel Platten vii PLAGIAT MERUPAKAN TINDAKAN TIDAK TERPUJI To my beloved parents, brothers, and sister with whom I share loves and hopes – one and only my destination viii PLAGIAT MERUPAKAN TINDAKAN TIDAK TERPUJI ACKNOWLEDGEMENTS First of all, I would like to say my gratitude to the only Almighty one, God. -

Adopt-An-Animal Program Thanks to These Groups and Individuals Who Participated in the Fort Wayne Children’S Zoo Adopt-An-Animal Program in 2013
Adopt-an-Animal Program Thanks to these groups and individuals who participated in the Fort Wayne Children’s Zoo Adopt-an-Animal program in 2013 A&M Logistical Services Inc. Adeline Jean Gosheen Liam Alexander Reynolds Reid Abbott Stan & Carol Gustin Kyle Richmond Michael Adams Kristen Haff Logan Richmond Alpha Omicron Pi Kappa Samantha Jane Hamilton Carolien Rietveld Kappa Chapter Kiley Anne Hatch Traci Rodriguez Estelle Armstrong Courtney Hause Christie Salcedo Alex Bailey Anne Higgins Jenna Sanders In Loving Memory of Ruby J. Teresa Hill Lee Sanga Bell Rod, Erica, Kyia, Karley and Jan & Steven Sarratore Vivian Blundall Khloe Hissong Chris & Monica Schiel Clara Blundall Doris Hook Kylee Shea Ashley Bollenbacher Hayden Page Hoover Mrs. Smith's First Grade Class Heidi Borgmann Chandler Hutchins Caela Jeanne Smith Madeline Borgmann Sarah C. Jesseph Olivia Smith Jennifer Boyd Julie Jesseph Audianna Snowden Zon Brain Balance Center of Fort Anna & Debbie Kirkpatrick Jill Solloway Wayne Mark Knirk Kimberly Spore Lily Brown Josalyn Kolberg Trinity Stephen Katie Buuck Samantha Kolberg Dona Stucko Anita Cast Nathan Kruger Brooke Thornhill Nathan Chamberlin Kasey LaFollette Dustin Tirpak Elijah Chapman Jenna Lane Diana VanderVelde Adrianna Clark Cathy R. Leugers Teri Voglewede Allison Clark KH luther Olivia Waggoner Anniston Lea Cooper Beth Marks Meagan Wallace Grandma & Grandpa Cox Patrick Masson Marci Wallace Avah Crane Lorene Meighan Melaina Walsh Dolores Crilly The Meuter Family Alexandria Wolf Amanda Cripps Lily M. Meyer George Wolf Keifer Cumings Stacey Miguel Ian Robert Yeaser Brandon Curry Emma Miller Mary Yurkanin William & Angela Davis Paige Misner Sara Zimmerman Hallie Mae Davis Catherine Moriarity Noah & Macy Rose from Gretchen Davis Clara Nonnenmacher Melinda Kuzdas Heidi Davis Faith Norem Julie DeBoer Caroline Odle We apologize if we failed to Addison Ebert Gail Overman Turner recognize your support. -

Degree Statute for the Master’S Degree Program
Degree Statute for the Master’s Degree Program Engineering Systems of HAN University of Applied Sciences academic year 2020 - 2021 Part 3 Education and Examination Regulations (EER) Content 1 Overview of the program.................................................................................................................. 3 2 Details of study: general modules ..................................................................................................... 5 Applied Control .................................................................................................................................... 6 Systems Modelling ............................................................................................................................. 23 3 Elective Modules ............................................................................................................................ 43 Advanced Controller Design ............................................................................................................... 44 Advanced Vehicle Dynamics ............................................................................................................... 55 Big Data & Small Data ........................................................................................................................ 67 Distributed Systems ........................................................................................................................... 78 Electric hybrid & fuel cell powertrains ............................................................................................... -

Name Clustering on the Basis of Parental Preferences Gerrit Bloothooft and Loek Groot Utrecht University, the Netherlands
names, Vol. 56, No. 3, September, 2008, 111–163 Name Clustering on the Basis of Parental Preferences Gerrit Bloothooft and Loek Groot Utrecht University, The Netherlands Parents do not choose fi rst names for their children at random. Using two large datasets, for the UK and the Netherlands, covering the names of children born in the same family over a period of two decades, this paper seeks to identify clusters of names entirely inferred from common parental naming preferences. These name groups can be considered as coherent sets of names that have a high probability to be found in the same family. Operational measures for the statistical association between names and clusters are developed, as well as a two-stage clustering technique. The name groups are subsequently merged into a limited set of grand clusters. The results show that clusters emerge with cultural, linguistic, or ethnic parental backgrounds, but also along characteristics inherent in names, such as clusters of names after fl owers and gems for girls, abbreviated names for boys, or names ending in –y or -ie. Introduction The variety in personal given names has increased enormously over the past century. In the Netherlands, the top 3, top 10, and top 100 names account, respectively, for 16%, 33%, and 70% of the fi rst names of elderly born between 1910 and 1930, while these fi gures are 3%, 8%, and 39% for babies born between 2000 and 2004. Compa- rable fi gures are presented by Galbi (2002, 4) for England and Wales. Along with the increase in the variety in names, the motives behind the choice of names for children by their parents have changed from a more or less prescribed naming after relatives to a free decision, a process that was facilitated in the Netherlands by the tolerant name law of 1970. -

M Oving M O Ving
MOVINGTOWARDS UNDERSTANDING:REASONING ABOUT GRAPHS IN PRIMARY MATHEMATICS EDUCATION MMOOVVINGING TOWARDS UNDERSTANDING CAROLIEN DUIJZER REASONINGREASONING ABOUT ABOUT GRAPHS GRAPHS IN PRIMARY IN MATHEMATICS EDUCATION REASONINGREASONING ABOUT ABOUT GRAPHS GRAPHS IN PRIMARY IN MATHEMATICS CAROLIEN EDUCATION DUIJZER PRIMARY MATHEMATICS EDUCATION CAROLIEN DUIJZER PRIMARY MATHEMATICS EDUCATION Moving towards understanding: Reasoning about graphs in primary mathematics education Carolien Duijzer 144737 Duijzer BNW.indd 1 30-07-20 12:03 Cover design and lay-out Carolien Duijzer Print Ridderprint, Alblasserdam ISBN: 978-94-6416-027-7 © 2020 Carolien Duijzer This research was carried out in the context of the Dutch Interuniversity Centre for Educational Research (ICO). The research reported in this dissertation was funded by the Netherlands Initiative for Education Research (Nationaal Regieorgaan Onderwijsonderzoek, NRO; projectnumber 405-14-303). All rights reserved. No part of this dissertation may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronically, mechanically, by photocopy, by recordings, or otherwise, without written permission from the author. 144737 Duijzer BNW.indd 2 30-07-20 12:03 Moving towards understanding: Reasoning about graphs in primary mathematics education Van beweging naar begrip: Redeneren over grafieken in het reken-wiskundeonderwijs (met een samenvatting in het Nederlands) Proefschrift ter verkrijging van de graad van doctor aan de Universiteit Utrecht op gezag van de rector magnificus, prof.dr. H.R.B.M. Kummeling, ingevolge het besluit van het college voor promoties in het openbaar te verdedigen op dinsdag 25 augustus 2020 des middags te 4.15 uur door Anneke Cornelia Gijsbertje Duijzer geboren op 24 januari 1989 te Brakel 144737 Duijzer BNW.indd 3 30-07-20 12:03 Promotoren: Prof. -

Chester County Marriages Grooms Index 1885-1930
Chester County Marriages Grooms Index 1885-1930 Groom's Last Name Groom's First Name Middle Name Groom's Date of Birth Groom's Age Bride's First Name Bride's Last Name Date of Application Date of Marriage Place of Marriage License # Tabb Lloyd 21 Sarah Sutton April 27, 1926 Phoenixville 26379 Tabb Lloyd 23 Annie Butts September 19, 1927 Phoenixville 27518 Taber Arthur M 27 Muriel Bennett November 8, 1924 West Chester 25431 Tabor Oliver KAugust 9, 1876 Mary Broomall October 1, 1904 West Chester 10635 Tackacs Andras 47 Maria Rendes February 10, 1921 22836 Taddeo Rinaldo 27 Michela DiPietrandonio September 6, 1914 Strafford 17856 Tafel LouisDecember 4, 1840 Anna Dutt June 27, 1886 Milltown 292 Taft Joseph 25 Hattie Daniels January 31, 1919 21239 Tagert GeorgeJuly 15, 1867 Lizzie Altemus July 29, 1891 Coatesville 2723 Taggart Earnest F 18 Sadie Brown February 11, 1918 Kelton 20620 Taggart Harold A 25 Isabelle Rettew September 1, 1920 Coatesville 22454 Taggart Harry EFebruary 18, 1887 Blanche Anderson March 11, 1910 Truce 14806 Taggart HowardDecember 8, 1861 Elizabeth Zimmermann October 5, 1892 Modena 3374 Taggart JamesApril 23, 1876 Meta Baehr September 6, 1905 Parkesburg 11306 Taggart John F 27 Bessie McHugh October 6, 1913 Phoenixville 17314 Taggart SamuelMay 16, 1880 Lena Gray May 18, 1902 Chatham 9102 Taggart Sylvester BMarch 20, 1857 Sarah Toland January 19, 1893 Sadsburyville 3541 Taggart Thomas A 22 Florence Hitman July 14, 1917 Phoenixville 20173 Taggart Thomas J 26 Mary Clooney June 14, 1917 Phoenixville 20063 Tailor John GilmoreSeptember 29, 1884 Sarah Keenan June 26, 1912 Phoenixville 16423 Tait CharlesJanuary 24, 1883 Esther Newcomer October 19, 1907 Lincoln University 12930 Tait Godfrey M. -

Bib Startlist Full IM 2907
Last update July 29th, 2021 BIB Age Group Surname First name AWA TriClub Country Represented 831 F18-24 Alen Bo BEL (Belgium) 1 F18-24 Gillespie Hannah GBR (United Kingdom of Great Britain and Northern Ireland) 442 F18-24 Haugen NiKoline NOR (Norway) 833 F18-24 Hughes Tess GBR (United Kingdom of Great Britain and Northern Ireland) 444 F18-24 Johansen Karoline FredriKstad TriatlonKlubb NOR (Norway) 1389 F18-24 Martin Hazel GBR (United Kingdom of Great Britain and Northern Ireland) 446 F18-24 Ryder Becca GBR (United Kingdom of Great Britain and Northern Ireland) 448 F25-29 ChetVeriKoVa Galina temptraining.ru RUS (Russian Federation) 3 F25-29 Circelli-Rauber Patricia AWA Gold JC Sports Coaching CHE (Switzerland) 1391 F25-29 DaValos Frida MEX (Mexico) 5 F25-29 De la Vega Fer MEX (Mexico) 1393 F25-29 Gonano Carolina ITA (Italy) 450 F25-29 HaaKana Anni FIN (Finland) 507 F25-29 Jaeger Cilia AWA Bronze DEU (Germany) 237 F25-29 Lysons Katie Clapham Chasers GBR (United Kingdom of Great Britain and Northern Ireland) 835 F25-29 Martin Sheona GBR (United Kingdom of Great Britain and Northern Ireland) 837 F25-29 OsmanKina Nadiia UKR (Ukraine) 1395 F25-29 Phillimore Paige GBR (United Kingdom of Great Britain and Northern Ireland) 246 F25-29 Rees Ffion GBR (United Kingdom of Great Britain and Northern Ireland) 452 F25-29 Sparrow Anna GBR (United Kingdom of Great Britain and Northern Ireland) 454 F25-29 Webb Tallulah GBR (United Kingdom of Great Britain and Northern Ireland) 509 F30-34 Ain el Fitre Nina AWA Bronze Triathlon Club Trigether CHE (Switzerland) 248 F30-34 Arfwidsson Autilia SWE (Sweden) 595 F30-34 Buddell Riin AWA SilVer EST (Estonia) 1397 F30-34 Cafri Noa MYWAY (ISRAEL) ISR (Israel) 239 F30-34 Cardoso Alice AWA Bronze BRA (Brazil) 241 F30-34 Christensen Isabella AWA SilVer DNK (DenmarK) 839 F30-34 Dr. -

1 Working Title: Menstruation and Menopause in Autistic Adults
Working title: Menstruation and Menopause in autistic adults: Periods of importance? Authors: Annabeth P. Groenman Carolien Torenvliet Tulsi A. Radhoe Joost A. Agelink van Rentergem Hilde M. Geurts Word count: Financial support: This work was funded by Innovational Research Incentives Scheme VICI (NWO) awarded to HMG (grant number 453-16-006). Declarations of interest: APG and CT, TAR, JAAR and HMG do not have any conflicts of interest to report 1 Abstract Autism spectrum conditions (ASC) were once seen as a predominantly male. This has caused a paucity of information on common events in the lives of women, such as menstruation and menopause. Some smaller studies indicate that autistic individuals might suffer from increased difficulties surrounding these events. This study aims to investigate, whether autistic individuals experience more frequent premenstrual dysphoric disorder (PMDD), and increased complaints surrounding menopause. In 70 individuals (ASC n=28, comparisons n= 42) we did not find an increased prevalence of PMDD in autistic individuals (14.3%) compared to non-autistic individuals (9.5%) . Autistic individuals 40 (n=75, ASC n=30, comparisons n= 35) did experience higher levels of menopausal complaints. In autistic individuals, higher menopausal complaints were associated with higher levels of depression and autistic traits. In non-autistic individuals, menopausal complaints were associated with increased inattention, hyperactivity/impulsivity (i.e., ADHD traits), and depression. With this work, we show the important role that major reproductive milestones can have in an autistic woman’s life. Lay abstract Autism spectrum conditions (ASC) were once seen as a predominantly male, but this has caused research to have little focus on women.