New Techniques of Laser Spectroscopy on Exotic Isotopes of Gallium and Francium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Point Defects in Lithium Gallate and Gallium Oxide
Air Force Institute of Technology AFIT Scholar Theses and Dissertations Student Graduate Works 8-23-2019 Point Defects in Lithium Gallate and Gallium Oxide Christopher A. Lenyk Follow this and additional works at: https://scholar.afit.edu/etd Part of the Nuclear Engineering Commons Recommended Citation Lenyk, Christopher A., "Point Defects in Lithium Gallate and Gallium Oxide" (2019). Theses and Dissertations. 2369. https://scholar.afit.edu/etd/2369 This Dissertation is brought to you for free and open access by the Student Graduate Works at AFIT Scholar. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of AFIT Scholar. For more information, please contact [email protected]. POINT DEFECTS IN LITHIUM GALLATE AND GALLIUM OXIDE DISSERTATION Christopher A. Lenyk, Lt Col, USAF AFIT-ENP-DS-19-S-023 DEPARTMENT OF THE AIR FORCE AIR UNIVERSITY AIR FORCE INSTITUTE OF TECHNOLOGY Wright-Patterson Air Force Base, Ohio DISTRIBUTION STATEMENT A. APPROVED FOR PUBLIC RELEASE; DISTRIBUTION UNLIMITED. The views expressed in this document are those of the author and do not reflect the official policy or position of the United States Air Force, the United States Department of Defense or the United States Government. This material is declared a work of the U.S. Government and is not subject to copyright protection in the United States. AFIT-ENP-DS-19-S-023 POINT DEFECTS IN LITHIUM GALLATE AND GALLIUM OXIDE DISSERTATION Presented to the Faculty Graduate School of Engineering and Management Air Force Institute of Technology Air University Air Education and Training Command in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Nuclear Engineering Christopher A. -

Z 221 PS2 Practice.Pages
CH 221 Practice Problem Set #2 This is a practice problem set and not the actual graded problem set that you will turn in for credit. Answers to each problem can be found at the end of this assignment. Covering: Chapter Two, Chapter 3.1 and Chapter Guide Two Important Tables and/or Constants: 1 mol = 6.022 x 1023 ! 1. Give the mass number of each of the following atoms: a. magnesium with 15 neutrons, b. titanium with 26 neutrons, and c. zinc with 32 neutrons. A 2. Give the complete symbol ( ZX) for each of the following atoms: a. potassium with 20 neutrons, b. krypton with 48 neutrons, and c. cobalt with 33 neutrons. 3. Thallium has two stable isotopes, 203Tl and 205Tl. Knowing that the atomic weight of thallium is 204.4, which isotope€ is the more abundant of the two? 4. Silver (Ag) has two stable isotopes, 107Ag and 109Ag. The isotopic mass of 107Ag is 106.9051 and the isotopic mass of 109Ag is 108.9047. The atomic weight of Ag, from the periodic table, is 107.868. Estimate the percentage of 107Ag in a sample of the element. a. 0% b. 25% c. 50% d. 75% 5. Gallium has two naturally occurring isotopes, 69Ga and 71Ga, with masses of 68.9257 u and 70.9249 u, respectively. Calculate the percent abundances of these isotopes of gallium. 6. Calculate the mass in grams of: a. 2.5 mol aluminum b. 1.25 x 10-3 mol of iron c. 0.015 mol of calcium d. -

Uncertainties in Cancer Risk Coefficients for Environmental Exposure to Radionuclides
OAK RIDGE ORNL/TM-2006/583 NATIONAL LABORATORY MANAGED BY UT-BATTELLE FOR THE DEPARTMENT OF ENERGY Uncertainties in Cancer Risk Coefficients for Environmental Exposure to Radionuclides An Uncertainty Analysis for Risk Coefficients Reported in Federal Guidance Report No. 13 January 2007 Prepared by D. J. Pawela R. W. Leggettb K. F. Eckermanb C. B. Nelsona aOffice of Radiation and Indoor Air U.S. Environmental Protection Agency Washington, DC 20460 bOak Ridge National Laboratory Oak Ridge, Tennessee 37831 DOCUMENT AVAILABILITY Reports produced after January 1, 1996, are generally available free via the U.S. Department of Energy (DOE) Information Bridge: Web site: http://www.osti.gov/bridge Reports produced before January 1, 1996, may be purchased by members of the public from the following source: National Technical Information Service 5285 Port Royal Road Springfield, VA 22161 Telephone: 703-605-6000 (1-800-553-6847) TDD: 703-487-4639 Fax: 703-605-6900 E-mail: [email protected] Web site: http://www.ntis.gov/support/ordernowabout.htm Reports are available to DOE employees, DOE contractors, Energy Technology Data Exchange (ETDE) representatives, and International Nuclear Information System (INIS) representatives from the following source: Office of Scientific and Technical Information P.O. Box 62 Oak Ridge, TN 37831 Telephone: 865-576-8401 Fax: 865-576-5728 E-mail: [email protected] Web site: http://www.osti.gov/contact.html This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. -

Isotope Geochemistry of Gallium in Hydrothermal Systems
ISOTOPE GEOCHEMISTRY OF GALLIUM IN HYDROTHERMAL SYSTEMS by Constance E. Payne A thesis submitted to Victoria University of Wellington in partial fulfilment of the requirements for the degree of Master of Science Victoria University of Wellington 2016 ABSTRACT Little is known about the isotope geochemistry of gallium in natural systems (Groot, 2009), with most information being limited to very early studies of gallium isotopes in extra-terrestrial samples (Aston, 1935; De Laeter, 1972; Inghram et al., 1948; Machlan et al., 1986). This study is designed as a reconnaissance for gallium isotope geochemistry in hydrothermal systems of New Zealand. Gallium has two stable isotopes, 69Ga and 71Ga, and only one oxidation state, Ga3+, in aqueous media (Kloo et al., 2002). This means that fractionation of gallium isotopes should not be effected by redox reactions. Therefore the physical processes that occur during phase changes of hydrothermal fluids (i.e. flashing of fluids to vapour phase and residual liquid phase) and mineralisation of hydrothermal precipitates (i.e. precipitation and ligand exchange) can be followed by studying the isotopes of gallium. A gallium anomaly is known to be associated with some hydrothermal processes as shown by the unusual, elevated concentrations (e.g. 290 ppm in sulfide samples of Waiotapu; this study) in several of the active geothermal systems in New Zealand. The gallium isotope system has not yet been investigated since the revolution of high precision isotopic ratio measurements by Multi-Collector Inductively Coupled Plasma Mass Spectrometry (MC-ICPMS) and so a new analytical methodology needed to be established. Any isotopic analysis of multi-isotope elements must satisfy a number of requirements in order for results to be both reliable and meaningful. -
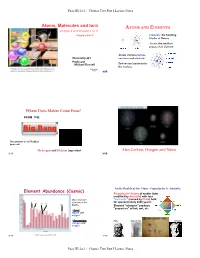
ATOMS and ELEMENTS Chapter 2 and Chapter 3 (3.1) “Chapter 2 Part 1” Elements: the Building Blocks of Nature Atoms: the Smallest Pieces of an Element
Page III-2a-1 / Chapter Two Part I Lecture Notes Atoms, Molecules and Ions ATOMS AND ELEMENTS Chapter 2 and Chapter 3 (3.1) “Chapter 2 Part 1” Elements: the building blocks of Nature Atoms: the smallest pieces of an element Atoms contain protons, Chemistry 221 neutrons and electrons Professor Michael Russell Protons and neutrons in the nucleus Last update: MAR 8/9/21 MAR Where Does Matter Come From? FROM THE Big Bang The universe is 13.77 billion years old Hydrogen and Helium important Also Carbon, Oxygen and Neon MAR MAR Early Models of the Atom - Empedocles & Aristotle Element Abundance (Cosmic) Empedocles’ theory of matter (later modified by Aristotle) with four Most abundant "elements" (coined by Plato) held C elements in the for approximately 2000 years! O Al Si Fe Earth's Element "mixtures" produce "properties" of hot, wet, etc. • Crust: Silicon and oxygen • Atmosphere: Plato Empedocles Aristotle nitrogen and oxygen MAR http://www.webelements.com/ MAR Page III-2a-1 / Chapter Two Part I Lecture Notes Page III-2a-2 / Chapter Two Part I Lecture Notes The "Newton" of Chemistry Early Models of the Atom - Democritus JOHN DALTON (1766 - 1844) DEMOCRITUS (460 - 370 BCE) 1804 - Proposed Atomic Theory was a contemporary of Plato "Atoms cannot be created or destroyed" "Atoms of one element are different from other Atoms have structure and volume element's atoms" - proposed relative scale of "Gold can be divided into atomic masses (now the amu) smaller pieces only so far before the pieces no longer "Chemical change involves bond breaking, retain -

Laser Spectroscopy of Gallium Isotopes Using the ISCOOL RFQ
Addendum to INTC-P-224 / IS457 Laser spectroscopy of gallium isotopes using the ISCOOL RFQ cooler. J. Billowes1, M.L. Bissell2, K. Blaum 3, B. Cheal1, K.T. Flanagan1, D.H. Forest4, C. Geppert5, M. Kowalska6, K. Kreim3, I.D. Moore7, R. Neugart8, G. Neyens2, W. N¨ortersh¨auser8, H.H. Stroke9, G. Tungate4, D.T. Yordanov6 1 Schuster Bldg, The University of Manchester, Brunswick Street, Manchester, M13 9PL, UK 2 Instituut voor Kern- en Stralingsfysica, KU Leuven, B-3001 Leuven, Belgium 3 Max-Planck-Institut f¨urKernphysik, D-69117 Heidelberg, Germany 4 School of Physics and Astronomy, The University of Birmingham, Birmingham, B15 2TT, UK 5 GSI Helmholtzzentrum f¨urSchwerionenforschung GmbH, D-64291 Darmstadt, Germany 6 Physics Department, CERN, CH-1211 Geneva 23, Switzerland 7 Department of Physics, University of Jyv¨askyl¨a,PB 35 (YFL) FIN-40014 Jyv¨askyl¨a,Finland 8 Institut f¨urKernchemie, Universit¨atMainz, D-55128 Mainz, Germany 9 Department of Physics, New York University, 4 Washington Place, New York, NY 10003, USA Spokesperson: Bradley Cheal Contact Person: Mark Bissell At the February 2007 meeting of the INTC, the above proposal was submitted to per- form laser spectroscopy of both neutron-rich and neutron-deficient isotopes of gallium using the newly installed ISCOOL. The INTC awarded 15 shifts for the study of the neutron-rich isotopes and requested that a status report then be submitted|discussing the measurements, performance of ISCOOL and beam purity|before proceeding to the neutron-deficient cases. The report is given here, with a request for 9 shifts to continue the study to the neutron-deficient isotopes. -

Development of Antibody Immuno-PET/SPECT Radiopharmaceuticals for Imaging of Oncological Disorders—An Update
cancers Review Development of Antibody Immuno-PET/SPECT Radiopharmaceuticals for Imaging of Oncological Disorders—An Update Jonatan Dewulf 1,3 , Karuna Adhikari 2, Christel Vangestel 1,3 , Tim Van Den Wyngaert 1,3 and Filipe Elvas 1,3,* 1 Molecular Imaging Center Antwerp, Faculty of Medicine and Health Sciences, University of Antwerp, Universiteitsplein 1, B-2610 Wilrijk, Belgium; [email protected] (J.D.); [email protected] (C.V.); [email protected] (T.V.D.W.) 2 Faculty of Pharmaceutical Biomedical and Veterinary Sciences, Medicinal Chemistry, University of Antwerp, Universiteitsplein 1, B-2610 Wilrijk, Belgium; [email protected] 3 Department of Nuclear Medicine, Antwerp University Hospital, Wilrijkstraat 10, B-2650 Edegem, Belgium * Correspondence: fi[email protected] Received: 8 June 2020; Accepted: 10 July 2020; Published: 11 July 2020 Abstract: Positron emission tomography (PET) and single-photon emission computed tomography (SPECT) are molecular imaging strategies that typically use radioactively labeled ligands to selectively visualize molecular targets. The nanomolar sensitivity of PET and SPECT combined with the high specificity and affinity of monoclonal antibodies have shown great potential in oncology imaging. Over the past decades a wide range of radio-isotopes have been developed into immuno-SPECT/PET imaging agents, made possible by novel conjugation strategies (e.g., site-specific labeling, click chemistry) and optimization and development of novel radiochemistry procedures. In addition, new strategies such as pretargeting and the use of antibody fragments have entered the field of immuno-PET/SPECT expanding the range of imaging applications. Non-invasive imaging techniques revealing tumor antigen biodistribution, expression and heterogeneity have the potential to contribute to disease diagnosis, therapy selection, patient stratification and therapy response prediction achieving personalized treatments for each patient and therefore assisting in clinical decision making. -

Exam I Answers
CHEM 1314 3;30 pm Theory Name ________________________ Exam !I John I. Gelder TA's Name ________________________ September 11, 2002 Lab Section ______________________ INSTRUCTIONS: 1. This examination consists of a total of 6 different pages. The last page include a periodic table and some useful equations. All work should be done in this booklet. 2. PRINT your name, TA's name and your lab section number now in the space at the top of this sheet. DO NOT SEPARATE THESE PAGES. 3. Answer all questions that you can and whenever called for show your work clearly. Your method of solving problems should pattern the approach used in lecture. 4. No credit will be awarded if your work is not shown in problems 2, 4 and 7a. 5. Point values are shown next to the problem number. 6. Budget your time for each of the questions. Some problems may have a low point value yet be very challenging. If you do not recognize the solution to a question quickly, skip it, and return to the question after completing the easier problems. 7. Look through the exam before beginning; plan your work; then begin. 8. Relax and do well. Page 2 Page 2 Page 3 Page 4 Page 5 TOTAL SCORES _____ _____ _____ _____ _____ ______ (1) (30) (24) (29) (16) (100) CHEM 1314 EXAM I PAGE 2 (12) 1. Write the chemical formula(s) of the product(s) and balance all of the following reactions. Identify all products phases as either (g)as, (l)iquid, (s)olid or (aq)ueous a) 3Mg(s) + N2(g) Æ Mg3N2(s) b) S8(g) + 8O2(g) Æ 8SO2(g) c) 2C6H14(l) + 19O2(g) Æ 12CO2(g) + 14H2O(g) d) 2K(s) + Cl2(g) Æ 2KCl(s) Grading: 3 points for the correct products, balance and phases. -

Chemistry 102 – Chapter 3 Summary and Review Answers
Chemistry 102 – Chapter 3 Summary and Review Answers 1. How are one mole of hydrogen gas and helium gas similar and how are they different? Answer: Within the context of Chapter 2 – one mole of hydrogen gas and one mole of helium gas contains the same number of gas particles. However, these particles for hydrogen are molecules (containing two atoms per molecule) and the particles for helium are atoms. Also, one mole of hydrogen molecules contains atoms with different composition and subsequently a different combined mass (2 g) compared to one mole of helium (4 g). 2. How are the states of matter denoted in a balanced chemical reaction? Answer: solid = s liquid = l gas = g aqueous = aq 3. Of formula units (or molecules), moles and mass – what can directly be compared for two different substances in a balanced chemical equation? Answer: Formula units and moles can be compared directly compared using stoichiometric coefficients from a balanced chemical equation. 4. Gallium has two stable isotopes, gallium‐69 and gallium‐71. If the mass of gallium‐69 is 68.925581 amu and is 60.108% abundant, what is the mass of gallium‐71? Answer: Because there are only two stable isotopes of gallium, the relative abundance of the two must combine to equal 100% or the relative abundance of gallium‐71 = 100% ‐ 60.81%. To organize the information: 69Ga 68.925581 amu 60.81% 71Ga to be determined 100% – 60.81% The average atomic mass is the weighted average of all of the stable isotopes: 69 71 69 69 ⎛⎞% Ga69 ⎛⎞ % Ga 71 ⎛⎞ % Ga 69 ⎛ 100%− % Ga ⎞ 71 Ga=+=+⎜⎟() Ga ⎜⎟() Ga ⎜⎟() Ga ⎜ ⎟() Ga ⎝⎠100% ⎝⎠ 100% ⎝⎠ 100% ⎝ 100% ⎠ Solving for the mass of gallium‐71: 69 ⎛⎞%Ga 69 ⎛⎞60.108% Ga− ⎜⎟() Ga 69.723 amu− ⎜⎟() 68.925581 amu 100% 100% 71Ga==⎝⎠ ⎝ ⎠ = 70.925 amu ⎛⎞100%− %69 Ga ⎛100%− 60.108% ⎞ ⎜⎟ ⎜⎟ ⎝⎠100% ⎝⎠100% 5. -

Foia/Pa-2015-0050
Acknowledgements The following were the members of ICRP Committee 2 who prepared this report. J. Vennart (Chairman); W. J. Bair; L. E. Feinendegen; Mary R. Ford; A. Kaul; C. W. Mays; J. C. Nenot; B. No∈ P. V. Ramzaev; C. R. Richmond; R. C. Thompson and N. Veall. The committee wishes to record its appreciation of the substantial amount of work undertaken by N. Adams and M. C. Thorne in the collection of data and preparation of this report, and, also, to thank P. E. Morrow, a former member of the committee, for his review of the data on inhaled radionuclides, and Joan Rowley for secretarial assistance, and invaluable help with the management of the data. The dosimetric calculations were undertaken by a task group, centred at the Oak Ridge Nntinnal-..._._.__. ---...“..,-J,1 .nhnmtnrv __ac ..,.I_.,“.fnllnwr~ Mary R. Ford (Chairwoman), S. R. Bernard, L. T. Dillman, K. F. Eckerman and Sarah B. Watson. Committee 2 wishes to record its indebtedness to the task group for the completion of this exacting task. The data given in this report are to be used together with the text and dosimetric models described in Part 1 of ICRP Publication 30;’ the chapters referred to in this preface relate to that report. In order to derive values of the Annual Limit on Intake (ALI) for radioisotopes of scandium, the following assumptions have been made. In the metabolic data for scandium, a fraction 0.4 of the element in the transfer compartment is translocated to the skeleton. It is assumed thit this scandium in the skeleton is distributed between cortical bone, trabecular bone and red marrow in proportion to their respective masses. -
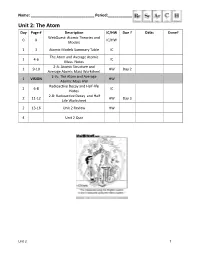
Unit 2: the Atom Day Page # Description IC/HW Due ? Date: Done? Webquest: Atomic Theories and 0 X IC/HW Models
Name: ______________________________ Period:_______________ Unit 2: The Atom Day Page # Description IC/HW Due ? Date: Done? WebQuest: Atomic Theories and 0 X IC/HW Models 1 3 Atomic Models Summary Table IC The Atom and Average Atomic 1 4-6 IC Mass Notes 2-A: Atomic Structure and 1 9-10 HW Day 2 Average Atomic Mass Worksheet 2-V1: The Atom and Average 1 VISION HW Atomic Mass HW Radioactive Decay and Half-life 2 6-8 IC Notes 2-B: Radioactive Decay and Half 2 11-12 HW Day 3 Life Worksheet 2 13-16 Unit 2 Review HW 4 Unit 2 Quiz Unit 2 1 Atomic Models Summary Table Name Characteristics Picture Dalton Electrons: (marble model) Protons: Neutrons: Other: Thomson Electrons: (plum pudding model) Protons: Neutrons: Other: Rutherford Electrons: (planetary model) Protons: Neutrons: Other: Bohr Electrons: (quantum model) Protons: Neutrons: Other: Important Scientists in the Atomic Theories and Models -atomos, initial idea of atom – Democritus - first atomic theory of matter, solid sphere model – John Dalton - discovery of the electron using the cathode ray tube experiment, plum pudding model – J. J. Thomson - discovery of the nucleus using the gold foil experiment, nuclear model – Ernest Rutherford - discovery of charge of electron using the oil drop experiment – Robert Millikan - energy levels, planetary model – Niels Bohr - periodic table arranged by atomic mass – Dmitri Mendeleev - periodic table arranged by atomic number – Henry Moseley - quantum nature of energy – Max Planck - uncertainty principle, quantum mechanical model – Werner Heisenberg -

(12) Patent Application Publication (10) Pub. No.: US 2004/0234450 A1 Howes (43) Pub
US 2004O234450A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2004/0234450 A1 HOWes (43) Pub. Date: Nov. 25, 2004 (54) COMPOSITIONS, METHODS, (60) Provisional application No. 60/262,635, filed on Jan. APPARATUSES, AND SYSTEMS FOR 22, 2001. SINGLET OXYGEN DELIVERY Publication Classification (76) Inventor: Randolph M. Howes, Kentwood, LA (US) (51) Int. Cl." ........................ A61K 51/00; A61M 36/14; A61K 33/40 Correspondence Address: (52) U.S. Cl. ........................................... 424/1.11; 424/616 CALFEE HALTER & GRISWOLD, LLP 800 SUPERIORAVENUE SUTE 1400 (57) ABSTRACT CLEVELAND, OH 44114 (US) (21) Appl. No.: 10/874,438 Methods of treating tumors, lesions, and cancers comprising delivering to the affected Site a combination of peroxide and (22) Filed: Jun. 23, 2004 hypochlorite anion. Hydrogen peroxide and Sodium hypochlorite are possible Sources of peroxide and hypochlo Related U.S. Application Data rite anion, respectively. The reactants may be injected Simul taneously or Sequentially, and combine at the Site to produce (60) Division of application No. 10/331,773, filed on Dec. Singlet oxygen. Singlet oxygen may be delivered to the 31, 2002, which is a continuation-in-part of applica treatment Site or generated at the treatment site. Isotopes are tion No. 10/050,121, filed on Jan. 18, 2002, which is also Synergistically used in conjunction with Singlet oxygen. a continuation-in-part of application No. 10/023,754, The isotopes may be radioactive isotopes, non-radioactive filed on Dec. 21, 2001. isotopes, or both. Patent Application Publication Nov. 25, 2004 Sheet 1 of 65 US 2004/0234450 A1 Fig.