The Global Translation Profile in a Ribosomal Protein Mutant Resembles That of an Eif3 Mutant
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Multiplexed Single-Cell Transcriptional Response Profiling to Define Cancer
ARTICLE https://doi.org/10.1038/s41467-020-17440-w OPEN Multiplexed single-cell transcriptional response profiling to define cancer vulnerabilities and therapeutic mechanism of action James M. McFarland 1,11, Brenton R. Paolella 1,11, Allison Warren1, Kathryn Geiger-Schuller 1,2, Tsukasa Shibue1, Michael Rothberg1, Olena Kuksenko1,2, William N. Colgan 1, Andrew Jones1, Emily Chambers1, Danielle Dionne1,2, Samantha Bender1, Brian M. Wolpin3,4,5, Mahmoud Ghandi 1, Itay Tirosh2,6, Orit Rozenblatt-Rosen1,2, Jennifer A. Roth1, Todd R. Golub 1,3,7,8, Aviv Regev 1,2,8,9,10, ✉ ✉ ✉ Andrew J. Aguirre 1,3,4,5,12 , Francisca Vazquez 1,12 & Aviad Tsherniak 1,12 1234567890():,; Assays to study cancer cell responses to pharmacologic or genetic perturbations are typically restricted to using simple phenotypic readouts such as proliferation rate. Information-rich assays, such as gene-expression profiling, have generally not permitted efficient profiling of a given perturbation across multiple cellular contexts. Here, we develop MIX-Seq, a method for multiplexed transcriptional profiling of post-perturbation responses across a mixture of samples with single-cell resolution, using SNP-based computational demultiplexing of single- cell RNA-sequencing data. We show that MIX-Seq can be used to profile responses to chemical or genetic perturbations across pools of 100 or more cancer cell lines. We combine it with Cell Hashing to further multiplex additional experimental conditions, such as post- treatment time points or drug doses. Analyzing the high-content readout of scRNA-seq reveals both shared and context-specific transcriptional response components that can identify drug mechanism of action and enable prediction of long-term cell viability from short- term transcriptional responses to treatment. -

Genes with 5' Terminal Oligopyrimidine Tracts Preferentially Escape Global Suppression of Translation by the SARS-Cov-2 NSP1 Protein
Downloaded from rnajournal.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Genes with 5′ terminal oligopyrimidine tracts preferentially escape global suppression of translation by the SARS-CoV-2 Nsp1 protein Shilpa Raoa, Ian Hoskinsa, Tori Tonna, P. Daniela Garciaa, Hakan Ozadama, Elif Sarinay Cenika, Can Cenika,1 a Department of Molecular Biosciences, University of Texas at Austin, Austin, TX 78712, USA 1Corresponding author: [email protected] Key words: SARS-CoV-2, Nsp1, MeTAFlow, translation, ribosome profiling, RNA-Seq, 5′ TOP, Ribo-Seq, gene expression 1 Downloaded from rnajournal.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract Viruses rely on the host translation machinery to synthesize their own proteins. Consequently, they have evolved varied mechanisms to co-opt host translation for their survival. SARS-CoV-2 relies on a non-structural protein, Nsp1, for shutting down host translation. However, it is currently unknown how viral proteins and host factors critical for viral replication can escape a global shutdown of host translation. Here, using a novel FACS-based assay called MeTAFlow, we report a dose-dependent reduction in both nascent protein synthesis and mRNA abundance in cells expressing Nsp1. We perform RNA-Seq and matched ribosome profiling experiments to identify gene-specific changes both at the mRNA expression and translation level. We discover that a functionally-coherent subset of human genes are preferentially translated in the context of Nsp1 expression. These genes include the translation machinery components, RNA binding proteins, and others important for viral pathogenicity. Importantly, we uncovered a remarkable enrichment of 5′ terminal oligo-pyrimidine (TOP) tracts among preferentially translated genes. -

Systematically Profiling the Expression of Eif3 Subunits in Glioma Reveals
Chai et al. Cancer Cell Int (2019) 19:155 https://doi.org/10.1186/s12935-019-0867-1 Cancer Cell International PRIMARY RESEARCH Open Access Systematically profling the expression of eIF3 subunits in glioma reveals the expression of eIF3i has prognostic value in IDH-mutant lower grade glioma Rui‑Chao Chai1,4,6†, Ning Wang2†, Yu‑Zhou Chang3, Ke‑Nan Zhang1,6, Jing‑Jun Li1,6, Jun‑Jie Niu5, Fan Wu1,6*, Yu‑Qing Liu1,6* and Yong‑Zhi Wang1,3,4,6* Abstract Background: Abnormal expression of the eukaryotic initiation factor 3 (eIF3) subunits plays critical roles in tumo‑ rigenesis and progression, and also has potential prognostic value in cancers. However, the expression and clinical implications of eIF3 subunits in glioma remain unknown. Methods: Expression data of eIF3 for patients with gliomas were obtained from the Chinese Glioma Genome Atlas (CGGA) (n 272) and The Cancer Genome Atlas (TCGA) (n 595). Cox regression, the receiver operating characteristic (ROC) curves= and Kaplan–Meier analysis were used to study= the prognostic value. Gene oncology (GO) and gene set enrichment analysis (GSEA) were utilized for functional prediction. Results: In both the CGGA and TCGA datasets, the expression levels of eIF3d, eIF3e, eIF3f, eIF3h and eIF3l highly were associated with the IDH mutant status of gliomas. The expression of eIF3b, eIF3i, eIF3k and eIF3m was increased with the tumor grade, and was associated with poorer overall survival [All Hazard ratio (HR) > 1 and P < 0.05]. By contrast, the expression of eIF3a and eIF3l was decreased in higher grade gliomas and was associated with better overall sur‑ vival (Both HR < 1 and P < 0.05). -

Translation Initiation Factor Eif3h Targets Specific Transcripts To
Translation initiation factor eIF3h targets specific transcripts to polysomes during embryogenesis Avik Choudhuria,b, Umadas Maitraa,1, and Todd Evansb,1 aDepartment of Developmental and Molecular Biology, Albert Einstein College of Medicine of Yeshiva University, Bronx, NY 10461; and bDepartment of Surgery, Weill Cornell Medical College, New York, NY 10065 Edited by Igor B. Dawid, The Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, and approved April 30, 2013 (received for review February 14, 2013) Eukaryotic translation initiation factor 3 (eIF3) plays a central role eukaryotes. These—eIF3d, eIF3e, eIF3f, eIF3h, eIF3j, eIF3k, in translation initiation and consists of five core (conserved) sub- eIF3l, and eIF3m—were designated “non-core” subunits (4). units present in both budding yeast and higher eukaryotes. Higher In contrast to the budding yeast, the genome of the fission eukaryotic eIF3 contains additional (noncore or nonconserved) yeast Schizosaccharomyces pombe contains structural homologs subunits of poorly defined function, including sub-unit h (eIF3h), of at least five noncore (nonconserved) eIF3 subunits—eIF3d, which in zebrafish is encoded by two distinct genes (eif3ha and eIF3e, eIF3f, eIF3h, and eIF3m. The gene encoding eIF3f is eif3hb). Previously we showed that eif3ha encodes the predominant essential for growth, whereas eIF3d, eIF3e, and eIF3h are dis- isoform during zebrafish embryogenesis and that depletion of this pensable for growth and viability (5–11). However, deleted factor causes defects in the development of the brain and eyes. To strains show specific phenotypes including defects in meiosis/ investigate the molecular mechanism governing this regulation, we sporulation (6, 9, 11). -

Identification of Differentially Expressed Genes and Pathways in Mice Exposed to Mixed Field Neutron/Photon Radiation Constantinos G
Broustas et al. BMC Genomics (2018) 19:504 https://doi.org/10.1186/s12864-018-4884-6 RESEARCHARTICLE Open Access Identification of differentially expressed genes and pathways in mice exposed to mixed field neutron/photon radiation Constantinos G. Broustas1* , Andrew D. Harken2, Guy Garty2 and Sally A. Amundson1 Abstract Background: Radiation exposure due to the detonation of an improvised nuclear device remains a major security concern. Radiation from such a device involves a combination of photons and neutrons. Although photons will make the greater contribution to the total dose, neutrons will certainly have an impact on the severity of the exposure as they have high relative biological effectiveness. Results: We investigated the gene expression signatures in the blood of mice exposed to 3 Gy x-rays, 0.75 Gy of neutrons, or to mixed field photon/neutron with the neutron fraction contributing 5, 15%, or 25% of a total 3 Gy radiation dose. Gene ontology and pathway analysis revealed that genes involved in protein ubiquitination pathways were significantly overrepresented in all radiation doses and qualities. On the other hand, eukaryotic initiation factor 2 (EIF2) signaling pathway was identified as one of the top 10 ranked canonical pathways in neutron, but not pure x-ray, exposures. In addition, the related mTOR and regulation of EIF4/p70S6K pathways were also significantly underrepresented in the exposures with a neutron component, but not in x-ray radiation. The majority of the changed genes in these pathways belonged to the ribosome biogenesis and translation machinery and included several translation initiation factors (e.g. Eif2ak4, Eif3f), as well as 40S and 60S ribosomal subunits (e.g. -

1 1 2 Pharmacological Dimerization and Activation of the Exchange
1 2 3 Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the 4 integrated stress response 5 6 7 *Carmela Sidrauski1,2, *Jordan C. Tsai1,2, Martin Kampmann2,3, Brian R. Hearn4, Punitha 8 Vedantham4, Priyadarshini Jaishankar4 , Masaaki Sokabe5, Aaron S. Mendez1,2, Billy W. 9 Newton6, Edward L. Tang6.7, Erik Verschueren6, Jeffrey R. Johnson6,7, Nevan J. Krogan6,7,, 10 Christopher S. Fraser5, Jonathan S. Weissman2,3, Adam R. Renslo4, and Peter Walter 1,2 11 12 1Department of Biochemistry and Biophysics, University of California, San Francisco, United 13 States 14 2Howard Hughes Medical Institute, University of California, San Francisco, United States 15 3Department of Cellular and Molecular Pharmacology, University of California, San Francisco, 16 United States 17 4Department of Pharmaceutical Chemistry and the Small Molecule Discovery Center, University 18 of California at San Francisco, United States 19 5Department of Molecular and Cellular Biology, College of Biological Sciences, University of 20 California, Davis, United States 21 6QB3, California Institute for Quantitative Biosciences, University of California, San Francisco, 22 United States 23 7Gladstone Institutes, San Francisco, United States 24 25 * Both authors contributed equally to this work 26 27 28 Abstract 29 30 The general translation initiation factor eIF2 is a major translational control point. Multiple 31 signaling pathways in the integrated stress response phosphorylate eIF2 serine-51, inhibiting 32 nucleotide exchange by eIF2B. ISRIB, a potent drug-like small molecule, renders cells 33 insensitive to eIF2α phosphorylation and enhances cognitive function in rodents by blocking 34 long-term depression. ISRIB was identified in a phenotypic cell-based screen, and its mechanism 35 of action remained unknown. -

Relevance of Translation Initiation in Diffuse Glioma Biology and Its
cells Review Relevance of Translation Initiation in Diffuse Glioma Biology and its Therapeutic Potential Digregorio Marina 1, Lombard Arnaud 1,2, Lumapat Paul Noel 1, Scholtes Felix 1,2, Rogister Bernard 1,3 and Coppieters Natacha 1,* 1 Laboratory of Nervous System Disorders and Therapy, GIGA-Neurosciences Research Centre, University of Liège, 4000 Liège, Belgium; [email protected] (D.M.); [email protected] (L.A.); [email protected] (L.P.N.); [email protected] (S.F.); [email protected] (R.B.) 2 Department of Neurosurgery, CHU of Liège, 4000 Liège, Belgium 3 Department of Neurology, CHU of Liège, 4000 Liège, Belgium * Correspondence: [email protected] Received: 18 October 2019; Accepted: 26 November 2019; Published: 29 November 2019 Abstract: Cancer cells are continually exposed to environmental stressors forcing them to adapt their protein production to survive. The translational machinery can be recruited by malignant cells to synthesize proteins required to promote their survival, even in times of high physiological and pathological stress. This phenomenon has been described in several cancers including in gliomas. Abnormal regulation of translation has encouraged the development of new therapeutics targeting the protein synthesis pathway. This approach could be meaningful for glioma given the fact that the median survival following diagnosis of the highest grade of glioma remains short despite current therapy. The identification of new targets for the development of novel therapeutics is therefore needed in order to improve this devastating overall survival rate. This review discusses current literature on translation in gliomas with a focus on the initiation step covering both the cap-dependent and cap-independent modes of initiation. -

Estrogenic Endocrine Disrupting Chemicals Influencing NRF1 Regulated Gene Networks in the Development of Complex Human Brain
Florida International University FIU Digital Commons All Faculty 12-13-2016 Estrogenic Endocrine Disrupting Chemicals Influencing NRF1 Regulated Gene Networks in the Development of Complex Human Brain Diseases Mark Preciados Department of Environmental & Occupational Health, Florida International University, [email protected] Changwon Yoo Department of Biostatistics, Florida International University, [email protected] Deodutta Roy Department of Environmental & Occupational Health, Florida International University, [email protected] Follow this and additional works at: https://digitalcommons.fiu.edu/all_faculty Recommended Citation Preciados, Mark; Yoo, Changwon; and Roy, Deodutta, "Estrogenic Endocrine Disrupting Chemicals Influencing NRF1 Regulated Gene Networks in the Development of Complex Human Brain Diseases" (2016). All Faculty. 180. https://digitalcommons.fiu.edu/all_faculty/180 This work is brought to you for free and open access by FIU Digital Commons. It has been accepted for inclusion in All Faculty by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. International Journal of Molecular Sciences Review Estrogenic Endocrine Disrupting Chemicals Influencing NRF1 Regulated Gene Networks in the Development of Complex Human Brain Diseases Mark Preciados 1, Changwon Yoo 2 and Deodutta Roy 1,* 1 Department of Environmental & Occupational Health, Florida International University, Miami, FL 33199, USA; mprec001@fiu.edu 2 Department of Biostatistics, Florida International University, Miami, FL 33199, USA; cyoo@fiu.edu * Correspondence: droy@fiu.edu; Tel.: +1-305-348-1694; Fax: +1-305-348-4901 Academic Editor: Paul B. Tchounwou Received: 25 October 2016; Accepted: 29 November 2016; Published: 13 December 2016 Abstract: During the development of an individual from a single cell to prenatal stages to adolescence to adulthood and through the complete life span, humans are exposed to countless environmental and stochastic factors, including estrogenic endocrine disrupting chemicals. -

New Partners Identified by Mass Spectrometry Assay Reveal Functions of NCAM2 in Neural Cytoskeleton Organization
International Journal of Molecular Sciences Article New Partners Identified by Mass Spectrometry Assay Reveal Functions of NCAM2 in Neural Cytoskeleton Organization Antoni Parcerisas 1,2,3,*,† , Alba Ortega-Gascó 1,2,† , Marc Hernaiz-Llorens 1,2 , Maria Antonia Odena 4, Fausto Ulloa 1,2, Eliandre de Oliveira 4, Miquel Bosch 3 , Lluís Pujadas 1,2 and Eduardo Soriano 1,2,* 1 Department of Cell Biology, Physiology and Immunology, University of Barcelona and Institute of Neurosciences, 08028 Barcelona, Spain; [email protected] (A.O.-G.); [email protected] (M.H.-L.); [email protected] (F.U.); [email protected] (L.P.) 2 Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED), 28031 Madrid, Spain 3 Department of Basic Sciences, Universitat Internacional de Catalunya, 08195 Sant Cugat del Vallès, Spain; [email protected] 4 Plataforma de Proteòmica, Parc Científic de Barcelona (PCB), 08028 Barcelona, Spain; [email protected] (M.A.O.); [email protected] (E.d.O.) * Correspondence: [email protected] (A.P.); [email protected] (E.S.) † A.P. and A.O.-G. contributed equally. Abstract: Neuronal cell adhesion molecule 2 (NCAM2) is a membrane protein with an important role in the morphological development of neurons. In the cortex and the hippocampus, NCAM2 is essential for proper neuronal differentiation, dendritic and axonal outgrowth and synapse forma- tion. However, little is known about NCAM2 functional mechanisms and its interactive partners Citation: Parcerisas, A.; during brain development. Here we used mass spectrometry to study the molecular interactome Ortega-Gascó, A.; Hernaiz-Llorens, of NCAM2 in the second postnatal week of the mouse cerebral cortex. -

This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: • This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. • A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. • This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. • The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. • When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Investigating the roles of Translation Elongation Factor 1B in mammalian cells Yuan Cao Thesis submitted for the degree of Doctor of Philosophy The University of Edinburgh 2012 Declaration I here declare that this thesis has been composed by me and that all the work presented within is my own, unless clearly indicated otherwise. This work has not been submitted for any other degree or professional qualification. Yuan Cao i Acknowledgements First of all I wish to express my deepest gratitude to my supervisor Professor Cathy Abbott who has been so patient and understanding in the last four years. I appreciate her scientific guidance throughout my research, her encouragement when I was being negative, as well as her support and help in both my work and life. -

(Eifs) in Endometrial Cancer
International Journal of Molecular Sciences Article The Prognostic Significance of Eukaryotic Translation Initiation Factors (eIFs) in Endometrial Cancer Maria Anna Smolle 1,2 , Piotr Czapiewski 3,4, Sylwia Lapi ´nska-Szumczyk 5, Hanna Majewska 3, Anna Supernat 6, Anna Zaczek 6 , Wojciech Biernat 3, Nicole Golob-Schwarzl 2,7 and Johannes Haybaeck 2,4,7,8,* 1 Department of Orthopaedics and Trauma, Medical University of Graz, Auenbruggerplatz 5, 8036 Graz, Austria; [email protected] 2 Area 2 Cancer, Center for Biomarker Research in Medicine, Stiftingtalstraße 5, 8010 Graz, Austria; [email protected] 3 Department of Pathomorphology, Medical University of Gdansk, Mariana Smoluchowskiego 17, 80-214 Gda´nsk,Poland; [email protected] (P.C.); [email protected] (H.M.); [email protected] (W.B.) 4 Department of Pathology, Medical Faculty, Otto-von-Guericke University Magdeburg, Leipziger Straße 44, 39120 Magdeburg, Germany 5 Department of Gynaecology, Gynaecological Oncology and Gynaecological Endocrinology, Medical University of Gda´nsk,M. Skłodowskiej-Curie 3a Street, 80-210 Gda´nsk,Poland; [email protected] 6 Department of Medical Biotechnology, Intercollegiate Faculty of Biotechnology, University of Gda´nnskand Medical University of Gda´nsk,Ba˙zy´nskiego1a, 80-952 Gda´nsk,Poland; [email protected] (A.S.); [email protected] (A.Z.) 7 Institute of Pathology, Medical University of Graz, Auenbruggerplatz 25, 8036 Graz, Austria 8 Department of Pathology, Neuropathology and Molecular Pathology, Medical University of Innsbruck, Müllerstraße 44, 6020 Innsbruck, Austria * Correspondence: [email protected]; Tel.: +49-391-67-15817 Received: 3 November 2019; Accepted: 5 December 2019; Published: 6 December 2019 Abstract: Whilst the role of eukaryotic translation initiation factors (eIFs) has already been investigated in several human cancers, their role in endometrial cancer (EC) is relatively unknown. -
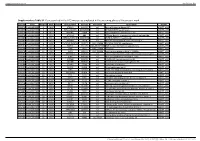
Supplementary Table S1. Genes Printed in the HC5 Microarray Employed in the Screening Phase of the Present Work
Supplementary material Ann Rheum Dis Supplementary Table S1. Genes printed in the HC5 microarray employed in the screening phase of the present work. CloneID Plate Position Well Length GeneSymbol GeneID Accession Description Vector 692672 HsxXG013989 2 B01 STK32A 202374 null serine/threonine kinase 32A pANT7_cGST 692675 HsxXG013989 3 C01 RPS10-NUDT3 100529239 null RPS10-NUDT3 readthrough pANT7_cGST 692678 HsxXG013989 4 D01 SPATA6L 55064 null spermatogenesis associated 6-like pANT7_cGST 692679 HsxXG013989 5 E01 ATP1A4 480 null ATPase, Na+/K+ transporting, alpha 4 polypeptide pANT7_cGST 692689 HsxXG013989 6 F01 ZNF816-ZNF321P 100529240 null ZNF816-ZNF321P readthrough pANT7_cGST 692691 HsxXG013989 7 G01 NKAIN1 79570 null Na+/K+ transporting ATPase interacting 1 pANT7_cGST 693155 HsxXG013989 8 H01 TNFSF12-TNFSF13 407977 NM_172089 TNFSF12-TNFSF13 readthrough pANT7_cGST 693161 HsxXG013989 9 A02 RAB12 201475 NM_001025300 RAB12, member RAS oncogene family pANT7_cGST 693169 HsxXG013989 10 B02 SYN1 6853 NM_133499 synapsin I pANT7_cGST 693176 HsxXG013989 11 C02 GJD3 125111 NM_152219 gap junction protein, delta 3, 31.9kDa pANT7_cGST 693181 HsxXG013989 12 D02 CHCHD10 400916 null coiled-coil-helix-coiled-coil-helix domain containing 10 pANT7_cGST 693184 HsxXG013989 13 E02 IDNK 414328 null idnK, gluconokinase homolog (E. coli) pANT7_cGST 693187 HsxXG013989 14 F02 LYPD6B 130576 null LY6/PLAUR domain containing 6B pANT7_cGST 693189 HsxXG013989 15 G02 C8orf86 389649 null chromosome 8 open reading frame 86 pANT7_cGST 693194 HsxXG013989 16 H02 CENPQ 55166