Embryonic Expression of the Common Progeroid Lamin a Splice Mutation Arrests Postnatal Skin Development
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Cell Line P53 Mutation Type UM
A Cell line p53 mutation Type UM-SCC 1 wt UM-SCC5 Exon 5, 157 GTC --> TTC Missense mutation by transversion (Valine --> Phenylalanine UM-SCC6 wt UM-SCC9 wt UM-SCC11A wt UM-SCC11B Exon 7, 242 TGC --> TCC Missense mutation by transversion (Cysteine --> Serine) UM-SCC22A Exon 6, 220 TAT --> TGT Missense mutation by transition (Tyrosine --> Cysteine) UM-SCC22B Exon 6, 220 TAT --> TGT Missense mutation by transition (Tyrosine --> Cysteine) UM-SCC38 Exon 5, 132 AAG --> AAT Missense mutation by transversion (Lysine --> Asparagine) UM-SCC46 Exon 8, 278 CCT --> CGT Missense mutation by transversion (Proline --> Alanine) B 1 Supplementary Methods Cell Lines and Cell Culture A panel of ten established HNSCC cell lines from the University of Michigan series (UM-SCC) was obtained from Dr. T. E. Carey at the University of Michigan, Ann Arbor, MI. The UM-SCC cell lines were derived from eight patients with SCC of the upper aerodigestive tract (supplemental Table 1). Patient age at tumor diagnosis ranged from 37 to 72 years. The cell lines selected were obtained from patients with stage I-IV tumors, distributed among oral, pharyngeal and laryngeal sites. All the patients had aggressive disease, with early recurrence and death within two years of therapy. Cell lines established from single isolates of a patient specimen are designated by a numeric designation, and where isolates from two time points or anatomical sites were obtained, the designation includes an alphabetical suffix (i.e., "A" or "B"). The cell lines were maintained in Eagle's minimal essential media supplemented with 10% fetal bovine serum and penicillin/streptomycin. -

Structural Heterogeneity of Cellular K5/K14 Filaments As Revealed by Cryo
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.12.442145; this version posted May 14, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Structural heterogeneity of cellular K5/K14 filaments as revealed by cryo- 2 electron microscopy 3 4 Short title: Structural heterogeneity of keratin filaments 5 6 7 Miriam S. Weber1, Matthias Eibauer1, Suganya Sivagurunathan2, Thomas M. Magin3, Robert D. 8 Goldman2, Ohad Medalia1* 9 1Department of Biochemistry, University of Zurich, Switzerland 10 2Department of Cell and Developmental Biology, Northwestern University Feinberg School of 11 Medicine, USA 12 3Institute of Biology, University of Leipzig, Germany 13 14 * Corresponding author: [email protected] 15 16 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.05.12.442145; this version posted May 14, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 17 Abstract 18 Keratin intermediate filaments are an essential and major component of the cytoskeleton in epithelial 19 cells. They form a stable yet dynamic filamentous network extending from the nucleus to the cell 20 periphery. Keratin filaments provide cellular resistance to mechanical stresses, ensure cell and tissue 21 integrity in addition to regulatory functions. Mutations in keratin genes are related to a variety of 22 epithelial tissue diseases that mostly affect skin and hair. Despite their importance, the molecular 23 structure of keratin filaments remains largely unknown. In this study, we analyzed the structure of 24 keratin 5/keratin 14 filaments within ghost keratinocytes by cryo-electron microscopy and cryo- 25 electron tomography. -

P63 Transcription Factor Regulates Nuclear Shape and Expression of Nuclear Envelope
Durham Research Online Deposited in DRO: 21 September 2017 Version of attached le: Published Version Peer-review status of attached le: Peer-reviewed Citation for published item: Rapisarda, Valentina and Malashchuk, Igor and Asamaowei, Inemo E. and Poterlowicz, Krzysztof and Fessing, Michael Y. and Sharov, Andrey A. and Karakesisoglou, Iakowos and Botchkarev, Vladimir A. and Mardaryev, Andrei (2017) 'p63 transcription factor regulates nuclear shape and expression of nuclear envelope-associated genes in epidermal keratinocytes.', Journal of investigative dermatology., 137 (10). pp. 2157-2167. Further information on publisher's website: https://doi.org/10.1016/j.jid.2017.05.013 Publisher's copyright statement: This article is available under the terms of the Creative Commons Attribution License (CC BY). You may copy and distribute the article, create extracts, abstracts and new works from the article, alter and revise the article, text or data mine the article and otherwise reuse the article commercially (including reuse and/or resale of the article) without permission from Elsevier. You must give appropriate credit to the original work, together with a link to the formal publication through the relevant DOI and a link to the Creative Commons user license above. You must indicate if any changes are made but not in any way that suggests the licensor endorses you or your use of the work. Additional information: Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in DRO • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. -
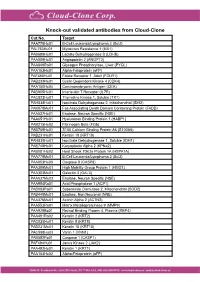
Knock-Out Validated Antibodies from Cloud-Clone Cat.No
Knock-out validated antibodies from Cloud-Clone Cat.No. Target PAA778Hu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAL763Hu01 Myxovirus Resistance 1 (MX1) PAB698Hu01 Lactate Dehydrogenase B (LDHB) PAA009Hu01 Angiopoietin 2 (ANGPT2) PAA849Ra01 Glycogen Phosphorylase, Liver (PYGL) PAA153Hu01 Alpha-Fetoprotein (aFP) PAF460Hu01 Folate Receptor 1, Adult (FOLR1) PAB233Hu01 Cyclin Dependent Kinase 4 (CDK4) PAA150Hu04 Carcinoembryonic Antigen (CEA) PAB905Hu01 Interleukin 7 Receptor (IL7R) PAC823Hu01 Thymidine Kinase 1, Soluble (TK1) PAH838Hu01 Isocitrate Dehydrogenase 2, mitochondrial (IDH2) PAK078Mu01 Fas Associating Death Domain Containing Protein (FADD) PAA537Hu01 Enolase, Neuron Specific (NSE) PAA651Hu01 Hyaluronan Binding Protein 1 (HABP1) PAB215Hu02 Fibrinogen Beta (FGb) PAB769Hu01 S100 Calcium Binding Protein A6 (S100A6) PAB231Hu01 Keratin 18 (KRT18) PAH839Hu01 Isocitrate Dehydrogenase 1, Soluble (IDH1) PAE748Hu01 Karyopherin Alpha 2 (KPNa2) PAB081Hu02 Heat Shock 70kDa Protein 1A (HSPA1A) PAA778Mu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAA853Hu03 Caspase 8 (CASP8) PAA399Mu01 High Mobility Group Protein 1 (HMG1) PAA303Mu01 Galectin 3 (GAL3) PAA537Mu02 Enolase, Neuron Specific (NSE) PAA994Ra01 Acid Phosphatase 1 (ACP1) PAB083Ra01 Superoxide Dismutase 2, Mitochondrial (SOD2) PAB449Mu01 Enolase, Non Neuronal (NNE) PAA376Mu01 Actinin Alpha 2 (ACTN2) PAA553Ra01 Matrix Metalloproteinase 9 (MMP9) PAA929Bo01 Retinol Binding Protein 4, Plasma (RBP4) PAA491Ra02 Keratin 2 (KRT2) PAC025Hu01 Keratin 8 (KRT8) PAB231Mu01 Keratin 18 (KRT18) PAC598Hu03 Vanin 1 (VNN1) -

Supplementary Material Contents
Supplementary Material Contents Immune modulating proteins identified from exosomal samples.....................................................................2 Figure S1: Overlap between exosomal and soluble proteomes.................................................................................... 4 Bacterial strains:..............................................................................................................................................4 Figure S2: Variability between subjects of effects of exosomes on BL21-lux growth.................................................... 5 Figure S3: Early effects of exosomes on growth of BL21 E. coli .................................................................................... 5 Figure S4: Exosomal Lysis............................................................................................................................................ 6 Figure S5: Effect of pH on exosomal action.................................................................................................................. 7 Figure S6: Effect of exosomes on growth of UPEC (pH = 6.5) suspended in exosome-depleted urine supernatant ....... 8 Effective exosomal concentration....................................................................................................................8 Figure S7: Sample constitution for luminometry experiments..................................................................................... 8 Figure S8: Determining effective concentration ......................................................................................................... -

Supplementary Figure 1
Supplementary Figure 1 A Cisplatin Cisplatin Cisplatin Late resistance Sensitive cells Early resistance (H357CisR8M) (H357CisS) (H357CisR4M) B SCC-4 CisS SCC9 CisS H357 CisS SCC-4 CisR 4M SCC9 CisR 4M 120 120 H357 CisR 4M SCC4 CisR 8M * H357 CisR 8M 120 SCC9 CisR 8M 100 * * * * 100 * * * * * 100 * * * * * * * * * * * * * * 80 * 80 * 80 60 60 60 40 40 40 % of cell viability of cell % % of cell viability of cell % 20 ofviability cell % 20 20 0 0 0 5 1 5 10 15 20 25 30 35 40 1 5 2.5 7.5 10 15 20 25 30 35 40 2.5 7.5 2.5 7.5 10 15 20 25 30 35 12.5 12.5 DMSO DMSO DMSO Cisplatin (M) Cisplatin (M) Cisplatin (M) C 100 100 80 80 60 60 40 A375 CisS 40 A549 CisS A549 CisR A375 CisR % of cell viability of cell % 20 % ofviability cell % 20 0 0 1 2 3 4 5 6 8 10 12 14 16 18 20 25 30 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 DMSO Cisplatin (M) DMSO Cisplatin (M) Supplementary figure 1: Characterization of sensitive, early and late cisplatin resistant lines: A) Schematic presentation of establishing sensitive, early and late cisplatin resistant cancer lines B) Sensitive, early and late cisplatin resistant pattern (CisS, CisR4M and CisR8M) of H357, SCC9 and SCC4 cells were treated with indicated concentrations of cisplatin for 48h and cell viability was determined by MTT assay (n=3, *: P < 0.05). C) Sensitive and late cisplatin resistant lung cancer (A549) and melanoma (A375) lines were established as described in method section. -

Keratin 1 Maintains Skin Integrity and Participates in an Inflammatory
Research Article 5269 Keratin 1 maintains skin integrity and participates in an inflammatory network in skin through interleukin-18 Wera Roth1, Vinod Kumar1, Hans-Dietmar Beer2, Miriam Richter1, Claudia Wohlenberg3, Ursula Reuter3, So¨ ren Thiering1, Andrea Staratschek-Jox4, Andrea Hofmann4, Fatima Kreusch4, Joachim L. Schultze4, Thomas Vogl5, Johannes Roth5, Julia Reichelt6, Ingrid Hausser7 and Thomas M. Magin1,* 1Translational Centre for Regenerative Medicine (TRM) and Institute of Biology, University of Leipzig, 04103 Leipzig, Germany 2University Hospital, Department of Dermatology, University of Zurich, 8006 Zurich, Switzerland 3Institute of Biochemistry and Molecular Biology, Division of Cell Biochemistry, University of Bonn, 53115 Bonn, Germany 4Department of Genomics and Immunoregulation, LIMES Institute, University of Bonn, 53115 Bonn, Germany 5Institute of Immunology, University of Mu¨nster, 48149 Mu¨nster, Germany 6Institute of Cellular Medicine and North East England Stem Cell Institute, Newcastle University, Newcastle upon Tyne NE2 4HH, UK 7Universita¨ts-Hautklinik, Ruprecht-Karls-Universita¨t Heidelberg, 69120 Heidelberg, Germany *Author for correspondence ([email protected]) Accepted 8 October 2012 Journal of Cell Science 125, 5269–5279 ß 2012. Published by The Company of Biologists Ltd doi: 10.1242/jcs.116574 Summary Keratin 1 (KRT1) and its heterodimer partner keratin 10 (KRT10) are major constituents of the intermediate filament cytoskeleton in suprabasal epidermis. KRT1 mutations cause epidermolytic ichthyosis in humans, characterized by loss of barrier integrity and recurrent erythema. In search of the largely unknown pathomechanisms and the role of keratins in barrier formation and inflammation control, we show here that Krt1 is crucial for maintenance of skin integrity and participates in an inflammatory network in murine keratinocytes. -

New Mesh Headings for 2018 Single Column After Cutover
New MeSH Headings for 2018 Listed in alphabetical order with Heading, Scope Note, Annotation (AN), and Tree Locations 2-Hydroxypropyl-beta-cyclodextrin Derivative of beta-cyclodextrin that is used as an excipient for steroid drugs and as a lipid chelator. Tree locations: beta-Cyclodextrins D04.345.103.333.500 D09.301.915.400.375.333.500 D09.698.365.855.400.375.333.500 AAA Domain An approximately 250 amino acid domain common to AAA ATPases and AAA Proteins. It consists of a highly conserved N-terminal P-Loop ATPase subdomain with an alpha-beta-alpha conformation, and a less-conserved C- terminal subdomain with an all alpha conformation. The N-terminal subdomain includes Walker A and Walker B motifs which function in ATP binding and hydrolysis. Tree locations: Amino Acid Motifs G02.111.570.820.709.275.500.913 AAA Proteins A large, highly conserved and functionally diverse superfamily of NTPases and nucleotide-binding proteins that are characterized by a conserved 200 to 250 amino acid nucleotide-binding and catalytic domain, the AAA+ module. They assemble into hexameric ring complexes that function in the energy-dependent remodeling of macromolecules. Members include ATPASES ASSOCIATED WITH DIVERSE CELLULAR ACTIVITIES. Tree locations: Acid Anhydride Hydrolases D08.811.277.040.013 Carrier Proteins D12.776.157.025 Abuse-Deterrent Formulations Drug formulations or delivery systems intended to discourage the abuse of CONTROLLED SUBSTANCES. These may include physical barriers to prevent chewing or crushing the drug; chemical barriers that prevent extraction of psychoactive ingredients; agonist-antagonist combinations to reduce euphoria associated with abuse; aversion, where controlled substances are combined with others that will produce an unpleasant effect if the patient manipulates the dosage form or exceeds the recommended dose; delivery systems that are resistant to abuse such as implants; or combinations of these methods. -

Mallory–Denk-Bodies: Lessons from Keratin-Containing Hepatic Inclusion Bodies
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1782 (2008) 764–774 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbadis Invited review Mallory–Denk-bodies: Lessons from keratin-containing hepatic inclusion bodies P. Strnad b, K. Zatloukal a, C. Stumptner a, H. Kulaksiz b, H. Denk a,⁎ a Institute of Pathology, Medical University of Graz, Auenbruggerplatz 25, A-8036 Graz, Austria b Department of Internal Medicine I, Center for Internal Medicine, University Medical Center Ulm, Albert-Einstein-Allee 23, D-89081 Ulm, Germany article info abstract Article history: Inclusion bodies are characteristic morphological features of various neuronal, muscular and other human Received 4 June 2008 disorders. They share common molecular constituents such as p62, chaperones and proteasome subunits. Received in revised form 25 August 2008 The proteins within aggregates are misfolded with increased β-sheet structure, they are heavily Accepted 26 August 2008 phosphorylated, ubiquitinylated and partially degraded. Furthermore, involvement of proteasomal system Available online 6 September 2008 represents a common feature of virtually all inclusions. Multiple aggregates contain intermediate filament proteins as their major constituents. Among them, Mallory–Denk bodies (MDBs) are the best studied. MDBs Keywords: Keratin represent hepatic inclusions observed in diverse chronic liver diseases such as alcoholic and non-alcoholic Mallory–Denk body steatohepatitis, chronic cholestasis, metabolic disorders and hepatocellular neoplasms. MDBs are induced in Aggregate mice fed griseofulvin or 3,5-diethoxycarbonyl-1,4-dihydrocollidine and resolve after discontinuation of toxin Inclusion administration. The availability of a drug-induced model makes MDBs a unique tool for studying inclusion Variant formation. -

Desmosome Structure, Composition and Function ⁎ David Garrod A, Martyn Chidgey B
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Available online at www.sciencedirect.com Biochimica et Biophysica Acta 1778 (2008) 572–587 www.elsevier.com/locate/bbamem Review Desmosome structure, composition and function ⁎ David Garrod a, Martyn Chidgey b, a Faculty of Life Sciences, University of Manchester, Michael Smith Building, Oxford Road, Manchester M13 9PT, UK b Division of Medical Sciences, University of Birmingham, Clinical Research Block, Queen Elizabeth Hospital, Birmingham B15 2TH, UK Received 24 May 2007; received in revised form 19 July 2007; accepted 20 July 2007 Available online 9 August 2007 Abstract Desmosomes are intercellular junctions of epithelia and cardiac muscle. They resist mechanical stress because they adopt a strongly adhesive state in which they are said to be hyper-adhesive and which distinguishes them from other intercellular junctions; desmosomes are specialised for strong adhesion and their failure can result in diseases of the skin and heart. They are also dynamic structures whose adhesiveness can switch between high and low affinity adhesive states during processes such as embryonic development and wound healing, the switching being signalled by protein kinase C. Desmosomes may also act as signalling centres, regulating the availability of signalling molecules and thereby participating in fundamental processes such as cell proliferation, differentiation and morphogenesis. Here we consider the structure, composition and function of desmosomes, and their role in embryonic development and disease. © 2007 Elsevier B.V. All rights reserved. Contents 1. Introduction .............................................................. 573 1.1. The strength of the desmosome–intermediate filament complex ................................ 573 1.2. Desmosomes resist mechanical stress because they are hyper-adhesive . -

In Silico Analysis Validates Proteomic Findings of Formalin-Fixed Paraffin Embedded Cutaneous Squamous Cell Carcinoma Tissue ALI AZIMI 1, KIMBERLEY L
CANCER GENOMICS & PROTEOMICS 13 : 453-466 (2016) doi:10.21873/cgp.20008 In Silico Analysis Validates Proteomic Findings of Formalin-fixed Paraffin Embedded Cutaneous Squamous Cell Carcinoma Tissue ALI AZIMI 1, KIMBERLEY L. KAUFMAN 2,3 , MARINA ALI 1, STEVEN KOSSARD 4 and PABLO FERNANDEZ-PENAS 1 1Department of Dermatology, Westmead Hospital, The University of Sydney, Westmead, NSW, Australia; 2School of Molecular Bioscience, Faculty of Science, The University of Sydney, Camperdown, NSW, Australia; 3Brain and Mind Centre, The University of Sydney, Camperdown, NSW, Australia; 4Dermatopathology, Skin and Cancer Foundation Australia, Darlinghurst, NSW, Australia Abstract. Background: Cutaneous squamous cell Cutaneous squamous cell carcinoma (cSCC) is a widespread carcinoma (cSCC) is a common type of skin cancer but there malignancy that is responsible for at least 20% of all non- are no comprehensive proteomic studies on this entity. melanoma skin cancer (NMSC) cases (1). The highest Materials and Methods: We employed liquid chromatography incidence of cSCC occurs in Australia, where a large coupled with tandem mass spectrometry (MS/MS) using Caucasian population has intense exposure to solar UV- formalin-fixed paraffin-embedded (FFPE) cSCC material to radiation (2, 3). In Australia from 1997 to 2010, NMSC study the tumor and normal skin tissue proteomes. Ingenuity treatments increased by 86%, and this number was projected Pathway Analysis (IPA) was used to interpret the role of to have increased a further 22% in 2015, with a total cost of altered proteins in cSCC pathophysiology. Results were AU $703.0 million for NMSC diagnosis and treatment (3). validated using the Human Protein Atlas and Oncomine While the majority of patients with early cSCC have a database in silico. -

Types I and II Keratin Intermediate Filaments
Downloaded from http://cshperspectives.cshlp.org/ on October 10, 2021 - Published by Cold Spring Harbor Laboratory Press Types I and II Keratin Intermediate Filaments Justin T. Jacob,1 Pierre A. Coulombe,1,2 Raymond Kwan,3 and M. Bishr Omary3,4 1Department of Biochemistry and Molecular Biology, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, Maryland 21205 2Departments of Biological Chemistry, Dermatology, and Oncology, School of Medicine, and Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University, Baltimore, Maryland 21205 3Departments of Molecular & Integrative Physiologyand Medicine, Universityof Michigan, Ann Arbor, Michigan 48109 4VA Ann Arbor Health Care System, Ann Arbor, Michigan 48105 Correspondence: [email protected] SUMMARY Keratins—types I and II—are the intermediate-filament-forming proteins expressed in epithe- lial cells. They are encoded by 54 evolutionarily conserved genes (28 type I, 26 type II) and regulated in a pairwise and tissue type–, differentiation-, and context-dependent manner. Here, we review how keratins serve multiple homeostatic and stress-triggered mechanical and nonmechanical functions, including maintenance of cellular integrity, regulation of cell growth and migration, and protection from apoptosis. These functions are tightly regulated by posttranslational modifications and keratin-associated proteins. Genetically determined alterations in keratin-coding sequences underlie highly penetrant and rare disorders whose pathophysiology reflects cell fragility or altered