Genetics and Epigenetics of Nicotine Dependence
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Birthdating of Myenteric Neuron Subtypes in the Small Intestine of the Mouse
RESEARCH ARTICLE Birthdating of Myenteric Neuron Subtypes in the Small Intestine of the Mouse Annette J. Bergner,1 Lincon A. Stamp,1 David G. Gonsalvez,1 Margaret B. Allison,2,3 David P. Olson,4 Martin G. Myers Jr,2,3,5 Colin R. Anderson,1 and Heather M. Young1* 1Department of Anatomy & Neuroscience, University of Melbourne, Victoria, Australia 2Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA 3Department of Molecular and Integrative Physiology, University of Michigan, Ann Arbor, Michigan, USA 4Division of Endocrinology, Department of Pediatrics, University of Michigan, Ann Arbor, Michigan, USA 5Department of Neuroscience Graduate Program, University of Michigan, Ann Arbor, Michigan, USA ABSTRACT vast majority of myenteric neurons had exited the cell There are many different types of enteric neurons. Pre- cycle by P10. We did not observe any EdU1/NOS11 vious studies have identified the time at which some myenteric neurons in the small intestine of adult mice enteric neuron subtypes are born (exit the cell cycle) in following EdU injection at E10.5 or E11.5, which was the mouse, but the birthdates of some major enteric unexpected, as previous studies have shown that NOS1 neuron subtypes are still incompletely characterized or neurons are present in E11.5 mice. Studies using the unknown. We combined 5-ethynynl-20-deoxyuridine proliferation marker Ki67 revealed that very few NOS1 (EdU) labeling with antibody markers that identify myen- neurons in the E11.5 and E12.5 gut were proliferating. teric neuron subtypes to determine when neuron sub- However, Cre-lox-based genetic fate-mapping revealed types are born in the mouse small intestine. -

The Baseline Structure of the Enteric Nervous System and Its Role in Parkinson’S Disease
life Review The Baseline Structure of the Enteric Nervous System and Its Role in Parkinson’s Disease Gianfranco Natale 1,2,* , Larisa Ryskalin 1 , Gabriele Morucci 1 , Gloria Lazzeri 1, Alessandro Frati 3,4 and Francesco Fornai 1,4 1 Department of Translational Research and New Technologies in Medicine and Surgery, University of Pisa, 56126 Pisa, Italy; [email protected] (L.R.); [email protected] (G.M.); [email protected] (G.L.); [email protected] (F.F.) 2 Museum of Human Anatomy “Filippo Civinini”, University of Pisa, 56126 Pisa, Italy 3 Neurosurgery Division, Human Neurosciences Department, Sapienza University of Rome, 00135 Rome, Italy; [email protected] 4 Istituto di Ricovero e Cura a Carattere Scientifico (I.R.C.C.S.) Neuromed, 86077 Pozzilli, Italy * Correspondence: [email protected] Abstract: The gastrointestinal (GI) tract is provided with a peculiar nervous network, known as the enteric nervous system (ENS), which is dedicated to the fine control of digestive functions. This forms a complex network, which includes several types of neurons, as well as glial cells. Despite extensive studies, a comprehensive classification of these neurons is still lacking. The complexity of ENS is magnified by a multiple control of the central nervous system, and bidirectional communication between various central nervous areas and the gut occurs. This lends substance to the complexity of the microbiota–gut–brain axis, which represents the network governing homeostasis through nervous, endocrine, immune, and metabolic pathways. The present manuscript is dedicated to Citation: Natale, G.; Ryskalin, L.; identifying various neuronal cytotypes belonging to ENS in baseline conditions. -

Nitric Oxide Enhances the Sensitivity of Alpaca Melanocytes to Respond to A-Melanocyte-Stimulating Hormone by Up-Regulating Melanocortin-1 Receptor
Biochemical and Biophysical Research Communications 396 (2010) 849–853 Contents lists available at ScienceDirect Biochemical and Biophysical Research Communications journal homepage: www.elsevier.com/locate/ybbrc Nitric oxide enhances the sensitivity of alpaca melanocytes to respond to a-melanocyte-stimulating hormone by up-regulating melanocortin-1 receptor Yanjun Dong, Jing Cao, Haidong Wang, Jie Zhang, Zhiwei Zhu, Rui Bai, HuanQing Hao, Xiaoyan He, Ruiwen Fan, Changsheng Dong * College of Animal Science and Technology, Shanxi Agricultural University, 030801 Taigu, Shanxi, China article info abstract Article history: Nitric oxide (NO) and a-melanocyte-stimulating hormone (a-MSH) have been correlated with the syn- Received 25 April 2010 thesis of melanin. The NO-dependent signaling of cellular response to activate the hypothalamopituitary Available online 6 May 2010 proopiomelanocortin system, thereby enhances the hypophysial secretion of a-MSH to stimulate a-MSH- receptor responsive cells. In this study we investigated whether an NO-induced pathway can enhance the Keywords: ability of the melanocyte to respond to a-MSH on melanogenesis in alpaca skin melanocytes in vitro.Itis Nitric oxide (NO) important for us to know how to enhance the coat color of alpaca. We set up three groups for experiments a-Melanocyte-stimulating hormone using the third passage number of alpaca melanocytes: the control cultures were allowed a total of 5 days (a-MSH) growth; the UV group cultures like the control group but the melanocytes were then irradiated everyday Melanocortin-1 receptor (MC1R) 2 Alpaca (once) with 312 mJ/cm of UVB; the UV + L-NAME group is the same as group UV but has the addition of Melanocyte 300 lM L-NAME (every 6 h). -

Influence of Functional Variant of Neuronal Nitric Oxide Synthase on Impulsive Behaviors in Humans
ORIGINAL ARTICLE Influence of Functional Variant of Neuronal Nitric Oxide Synthase on Impulsive Behaviors in Humans Andreas Reif, MD; Christian P. Jacob, MD; Dan Rujescu, MD; Sabine Herterich, PhD; Sebastian Lang, MD; Lise Gutknecht, PhD; Christina G. Baehne, Dipl-Psych; Alexander Strobel, PhD; Christine M. Freitag, MD; Ina Giegling, MD; Marcel Romanos, MD; Annette Hartmann, MD; Michael Rösler, MD; Tobias J. Renner, MD; Andreas J. Fallgatter, MD; Wolfgang Retz, MD; Ann-Christine Ehlis, PhD; Klaus-Peter Lesch, MD Context: Human personality is characterized by sub- Main Outcome Measures: For the association stud- stantial heritability but few functional gene variants have ies, the major outcome criteria were phenotypes rel- been identified. Although rodent data suggest that the evant to impulsivity, namely, the dimensional pheno- neuronal isoform of nitric oxide synthase (NOS-I) modi- type conscientiousness and the categorical phenotypes fies diverse behaviors including aggression, this has not adult ADHD, aggression, and cluster B personality been translated to human studies. disorder. Objectives: To investigate the functionality of an NOS1 Results: A novel functional promoter polymorphism in promoter repeat length variation (NOS1 Ex1f variable NOS1 was associated with traits related to impulsivity, number tandem repeat [VNTR]) and to test whether it including hyperactive and aggressive behaviors. Specifi- is associated with phenotypes relevant to impulsivity. cally, the short repeat variant was more frequent in adult ADHD, cluster B personality disorder, and autoaggres- Design: Molecular biological studies assessed the cel- sive and heteroaggressive behavior. This short variant lular consequences of NOS1 Ex1f VNTR; association came along with decreased transcriptional activity of the studies were conducted to investigate the impact of this genetic variant on impulsivity; imaging genetics was ap- NOS1 exon 1f promoter and alterations in the neuronal plied to determine whether the polymorphism is func- transcriptome including RGS4 and GRIN1. -

The Genetics of Bipolar Disorder
Molecular Psychiatry (2008) 13, 742–771 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp FEATURE REVIEW The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions A Serretti and L Mandelli Institute of Psychiatry, University of Bologna, Bologna, Italy Bipolar disorder (BP) is a complex disorder caused by a number of liability genes interacting with the environment. In recent years, a large number of linkage and association studies have been conducted producing an extremely large number of findings often not replicated or partially replicated. Further, results from linkage and association studies are not always easily comparable. Unfortunately, at present a comprehensive coverage of available evidence is still lacking. In the present paper, we summarized results obtained from both linkage and association studies in BP. Further, we indicated new potential interesting genes, located in genome ‘hot regions’ for BP and being expressed in the brain. We reviewed published studies on the subject till December 2007. We precisely localized regions where positive linkage has been found, by the NCBI Map viewer (http://www.ncbi.nlm.nih.gov/mapview/); further, we identified genes located in interesting areas and expressed in the brain, by the Entrez gene, Unigene databases (http://www.ncbi.nlm.nih.gov/entrez/) and Human Protein Reference Database (http://www.hprd.org); these genes could be of interest in future investigations. The review of association studies gave interesting results, as a number of genes seem to be definitively involved in BP, such as SLC6A4, TPH2, DRD4, SLC6A3, DAOA, DTNBP1, NRG1, DISC1 and BDNF. -

Competitive Metabolism of L-Arginine: Arginase As a Therapeutic Target In
JBR Journal of Biomedical Research,2011,25(5):299-308 http://elsevier.com/wps/ Review find/journaldescription.cws_ home/723905/description#description Competitive metabolism of L-arginine: arginase as a therapeutic ☆ target in asthma * Jennifer M. Bratt, Amir A. Zeki, Jerold A. Last, Nicholas J. Kenyon Department of Internal Medicine, Division of Pulmonary and Critical Care and Sleep Medicine, University of California, Davis, CA 95616, USA Received 25 April 2011, Revised 24 June 2011, Accepted 21 July 2011 Abstract Exhaled breath nitric oxide (NO) is an accepted asthma biomarker. Lung concentrations of NO and its amino acid precursor, L-arginine, are regulated by the relative expressions of the NO synthase (NOS) and arginase iso- forms. Increased expression of arginase I and NOS2 occurs in murine models of allergic asthma and in biopsies of asthmatic airways. Although clinical trials involving the inhibition of NO-producing enzymes have shown mixed results, small molecule arginase inhibitors have shown potential as a therapeutic intervention in animal and cell culture models. Their transition to clinical trials is hampered by concerns regarding their safety and potential tox- icity. In this review, we discuss the paradigm of arginase and NOS competition for their substrate L-arginine in the asthmatic airway. We address the functional role of L-arginine in inflammation and the potential role of arginase inhibitors as therapeutics. Keywords: nitric oxide, L-arginine, arginase, nor-NOHA, nitrosation, nitric oxide synthase INTRODUCTION from accumulation of collagens in the submucosal and [3] Asthma is a common disease characterized by a reticular basement membrane . The airway remod- syndrome of persistent airway inflammation and re- eling and resultant reduction in overall lung function versible airway obstruction. -

Three Classes of Tetrahydrobiopterin-Dependent Enzymes
DOI 10.1515/pterid-2013-0003 Pteridines 2013; 24(1): 7–11 Review Ernst R. Werner* Three classes of tetrahydrobiopterin-dependent enzymes Abstract: Current knowledge distinguishes three classes in Antalya, Turkey. For a more detailed review on bio- of tetrahydrobiopterin-dependent enzymes as based on chemistry and pathophysiology of tetrahydrobio pterin, protein sequence similarity. These three protein sequence the reader is referred to Ref. [ 1 ]. Here, a short historical clusters hydroxylate three types of substrate atoms and overview of the discovery of tetrahydrobiopterin-depend- use three different forms of iron for catalysis. The first ent enzymes is presented, followed by a summary of the class to be discovered was the aromatic amino acid biochemical properties of these enzymes. The reactions hydroxylases, which, in mammals, include phenylalanine catalyzed by the three classes of tetrahydrobiopterin- hydroxylase, tyrosine hydroxylase, and two isoforms of dependent enzymes are detailed in Figure 1 . tryptophan hydroxylases. The protein sequences of these tetrahydrobiopterin-dependent aromatic amino acid hydroxylases are significantly similar, and all mammalian Discovery of tetrahydrobiopterin- aromatic amino acid hydroxylases require a non-heme- dependent enzymes bound iron atom in the active site of the enzyme for cataly- sis. The second classes of tetrahydrobiopterin-dependent enzymes to be characterized were the nitric oxide syn- Aromatic amino acid hydroxylases thases, which in mammals occur as three isoforms. Nitric oxide synthase protein sequences form a separate cluster Phenylalanine hydroxylase was the first enzyme charac- of homologous sequences with no similarity to aromatic terized to be dependent on a tetrahydropterin [ 2 ]. It then amino acid hydroxylase protein sequences. In contrast to took five more years to identify the nature of the endo- aromatic amino acid hydroxylases, nitric oxide synthases genous cofactor as tetrahydrobiopterin [ 3 ]. -

Cellular Distribution and Function of Soluble Guanylyl Cyclase in Rat Kidney and Liver
ARTICLES J Am Soc Nephrol 12: 2209–2220, 2001 Cellular Distribution and Function of Soluble Guanylyl Cyclase in Rat Kidney and Liver FRANZISKA THEILIG,* MAGDALENA BOSTANJOGLO,* HERMANN PAVENSTADT,¨ † CLEMENS GRUPP,‡ GUDRUN HOLLAND,* ILKA SLOSAREK,* AXEL M. GRESSNER,§ MICHAEL RUSSWURM, DORIS KOESLING, and SEBASTIAN BACHMANN* *Department of Anatomy, Charite´, Humboldt University, Berlin, Germany; †Department of Nephrology, University of Freiburg, Freiburg, Germany; ‡Department of Nephrology, University of Go¨ttingen, Go¨ttingen, Germany; §Department of Clinical Chemistry, Rheinisch-Westfa¨lische Technische Hochschule Aachen, Aachen, Germany; and Department of Pharmacology, Ruhr University, Bochum, Germany. Abstract. Soluble guanylyl cyclase (sGC) catalyzes the biosyn- pendent accumulation of cGMP in cytosolic extracts of tissues thesis of cGMP in response to binding of L-arginine-derived and cells was measured in vitro. Renal glomerular arterioles, nitric oxide (NO). Functionally, the NO-sGC-cGMP signaling including the renin-producing granular cells, mesangium, and pathway in kidney and liver has been associated with regional descending vasa recta, as well as cortical and medullary inter- hemodynamics and the regulation of glomerular parameters. stitial fibroblasts, expressed sGC. Stimulation of isolated mes- The distribution of the ubiquitous sGC isoform ␣11 sGC was angial cells, renal fibroblasts, and hepatic Ito cells with a NO studied with a novel, highly specific antibody against the 1 donor resulted in markedly increased cytosolic cGMP levels. subunit. In parallel, the presence of mRNA encoding both This assessment of sGC expression and activity in vascular and subunits was investigated by using in situ hybridization and interstitial cells of kidney and liver may have implications for reverse transcription-PCR assays. -

Nos1-Adaptor Protein Dysfunction in the Nucleus Tractus Solitarii Contributes to the Neurogenic Heart Damage and Qt Interval Prolongation
NOS1-ADAPTOR PROTEIN DYSFUNCTION IN THE NUCLEUS TRACTUS SOLITARII CONTRIBUTES TO THE NEUROGENIC HEART DAMAGE AND QT INTERVAL PROLONGATION A Dissertation Submitted to the Graduate Faculty of the North Dakota State University of Agriculture and Applied Science By Neha Singh In Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Department: Cellular and Molecular Biology November 2014 Fargo, North Dakota North Dakota State University Graduate School Title NOS1-ADAPTOR PROTEIN DYSFUNCTION IN THE NUCLEUS TRACTUS SOLITARII CONTRIBUTES TO THE NEUROGENIC HEART DAMAGE AND QT INTERVAL PROLONGATION By Neha Singh The Supervisory Committee certifies that this disquisition complies with North Dakota State University’s regulations and meets the accepted standards for the degree of DOCTOR OF PHILOSOPHY SUPERVISORY COMMITTEE: Chengwen Sun, M.D., Ph.D. Chair Stephen T. O’Rourke, Ph.D. Jagdish Singh, Ph.D. Mark Sheridan, Ph.D. Approved: 4/20/15 Jane Schuh, Ph.D. Date Department Chair ABSTRACT Variants of the Nitric Oxide Synthase 1 Adaptor Protein (NOS1AP) locus are strongly related to QT interval prolongation and sudden cardiac death (SCD) in human. Neurogenic cardiac damage due to subarachnoid hemorrhage, stroke, epilepsy and myocardial infarction is known to contribute to sudden death in most cases. Our aim was to study the role of NOS1AP in the neurogenic cardiac damage by silencing NOS1AP expression in the Nucleus Tractus Solitarii (NTS) area of the brainstem using lentiviral vector-mediated NOS1AP shRNA (Lv-NOS1AP- shRNA). Real time PCR data showed NOS1AP mRNA levels were expressed in the NTS 3-fold higher than other organs such as kidney and heart in Sprague Dawley (SD) rats. -
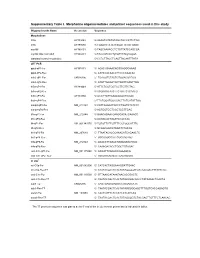
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC -
Arginase: Marker, Effector, Or Candidate Gene for Asthma?
Arginase: marker, effector, or candidate gene for asthma? Donata Vercelli J Clin Invest. 2003;111(12):1815-1817. https://doi.org/10.1172/JCI18908. Commentary Microarray analysis of the expression profiles of lung tissue in two murine models of asthma revealed high levels of arginase I and arginase II activity, in association with IL-4 and IL-13 overexpression, suggesting that arginine pathways are critical in the pathogenesis of asthma. Find the latest version: https://jci.me/18908/pdf molecular bases of the TNF signaling. initiation in the absence of caspase-8. J. Biol. Chem. is a direct inhibitor of apoptosis signal- regulat- 276:46639–46646. ing kinase (ASK) 1. EMBO J. 17:2596–2606. Given its important contribution to 5. Wajant, H., and Scheurich, P. 2001. Tumor necro- 13. Zhang, L., Chen, J., and Fu, H. 1999. Suppression TNF-induced JNK activation, AIP1 sis factor receptor-associated factor (TRAF) 2 and of apoptosis signal-regulating kinase 1-induced may represent a suitable target for pos- its role in TNF signaling. Int. J. Biochem. Cell. Biol. cell death by 14-3-3 proteins. Proc. Natl. Acad. Sci. 33:19–32. U. S. A. 96:8511–8515. sible therapeutic applications in 6. Ip, Y.T., and Davis, R.J. 1998. Signal transduction 14. Zhang, R., et al. 2003. AIP1 mediates TNF-α– human diseases characterized by by the c-jun N-terminal kinase (JNK): from induced ASK1 activation by facilitating disso- inflammation to development. Curr. Opin. Cell ciation of ASK1 from its inhibitor 14-3-3. increased TNF-α–mediated apoptosis. -

Hypoxia Induces a Functionally Significant and Translationally Efficient Neuronal NO Synthase Mrna Variant
Hypoxia induces a functionally significant and translationally efficient neuronal NO synthase mRNA variant Michael E. Ward, … , Derek C. Newton, Philip A. Marsden J Clin Invest. 2005;115(11):3128-3139. https://doi.org/10.1172/JCI20806. Research Article Vascular biology We tested the hypothesis that induction of neuronal NO synthase (nNOS) impairs vascular smooth muscle contractility after hypoxia. nNOS protein was increased in aorta, mesenteric arterioles, pulmonary arteries, brain, and diaphragm from 2+ rats exposed to 8% O2 for 48 hours and in human aortic SMCs after hypoxic incubation (1% O2). Ca -dependent NO synthase activity was increased in endothelium-denuded aortic segments from hypoxia-exposed rats. NG-nitro-L-arginine methyl ester enhanced the contractile responses of endothelium-denuded aortic rings and mesenteric arterioles from hypoxia-exposed but not normoxic rats (P < 0.05). The hypoxia-inducible mRNA transcript expressed by human cells was found to contain a novel 5′-untranslated region, consistent with activation of transcription in the genomic region contiguous with exon 2. Translational efficiency of this transcript is markedly increased compared with previously described human nNOS mRNAs. Transgenic mice possessing a lacZ reporter construct under control of these genomic sequences demonstrated expression of the construct after exposure to hypoxia (8% O2, 48 hours) in the aorta, mesenteric arterioles, renal papilla, and brain. These results reveal a novel human nNOS promoter that confers the ability to rapidly upregulate nNOS expression in response to hypoxia with a functionally significant effect on vascular smooth muscle contraction. Find the latest version: https://jci.me/20806/pdf Research article Related Commentary, page 2976 Hypoxia induces a functionally significant and translationally efficient neuronal NO synthase mRNA variant Michael E.