A Heterozygous Variant in the SLC22A12 Gene in a Sri Lanka
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evaluation of Abnormal Liver Chemistries
ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries Paul Y. Kwo, MD, FACG, FAASLD1, Stanley M. Cohen, MD, FACG, FAASLD2, and Joseph K. Lim, MD, FACG, FAASLD3 1Division of Gastroenterology/Hepatology, Department of Medicine, Stanford University School of Medicine, Palo Alto, California, USA; 2Digestive Health Institute, University Hospitals Cleveland Medical Center and Division of Gastroenterology and Liver Disease, Department of Medicine, Case Western Reserve University School of Medicine, Cleveland, Ohio, USA; 3Yale Viral Hepatitis Program, Yale University School of Medicine, New Haven, Connecticut, USA. Am J Gastroenterol 2017; 112:18–35; doi:10.1038/ajg.2016.517; published online 20 December 2016 Abstract Clinicians are required to assess abnormal liver chemistries on a daily basis. The most common liver chemistries ordered are serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase and bilirubin. These tests should be termed liver chemistries or liver tests. Hepatocellular injury is defined as disproportionate elevation of AST and ALT levels compared with alkaline phosphatase levels. Cholestatic injury is defined as disproportionate elevation of alkaline phosphatase level as compared with AST and ALT levels. The majority of bilirubin circulates as unconjugated bilirubin and an elevated conjugated bilirubin implies hepatocellular disease or cholestasis. Multiple studies have demonstrated that the presence of an elevated ALT has been associated with increased liver-related mortality. A true healthy normal ALT level ranges from 29 to 33 IU/l for males, 19 to 25 IU/l for females and levels above this should be assessed. The degree of elevation of ALT and or AST in the clinical setting helps guide the evaluation. -

Total Bilirubin in Athletes, Determination of Reference Range
OriginalTotal bilirubin Paper in athletes DOI: 10.5114/biolsport.2017.63732 Biol. Sport 2017;34:45-48 Total bilirubin in athletes, determination of reference range AUTHORS: Witek K1, Ścisłowska J2, Turowski D1, Lerczak K1, Lewandowska-Pachecka S2, Corresponding author: Pokrywka A3 Konrad Witek Department of Biochemistry, Institute of Sport - National 1 Department of Biochemistry, Institute of Sport - National Research Institute, Warsaw, Poland Research Institute 2 01-982 Warsaw, Poland Faculty of Pharmacy with the Laboratory Medicine Division, Medical University of Warsaw, Poland 3 E-mail: Department of Biochemistry, 2nd Faculty of Medicine, Medical University of Warsaw, Poland [email protected] ABSTRACT: The purpose of this study was to determine a typical reference range for the population of athletes. Results of blood tests of 339 athletes (82 women and 257 men, aged 18-37 years) were retrospectively analysed. The subjects were representatives of different sports disciplines. The measurements of total bilirubin (BIT), iron (Fe), alkaline phosphatase (ALP), alanine aminotransferase (ALT) and gamma-glutamyltransferase (GGT) were made using a Pentra 400 biochemical analyser (Horiba, France). Red blood cell count (RBC), reticulocyte count and haemoglobin concentration measurements were made using an Advia 120 haematology analyser (Siemens, Germany). In groups of women and men the percentage of elevated results were similar at 18%. Most results of total bilirubin in both sexes were in the range 7-14 μmol ∙ L-1 (49% of women and 42% of men). The highest results of elevated levels of BIT were in the range 21-28 μmol ∙ L-1 (12% of women and 11% of men). There was a significant correlation between serum iron and BIT concentration in female and male athletes whose serum total bilirubin concentration does not exceed the upper limit of the reference range. -

Viewed Under 23 (B) Or 203 (C) fi M M Male Cko Mice, and Largely Unaffected Magni Cation; Scale Bars, 500 M (B) and 50 M (C)
BRIEF COMMUNICATION www.jasn.org Renal Fanconi Syndrome and Hypophosphatemic Rickets in the Absence of Xenotropic and Polytropic Retroviral Receptor in the Nephron Camille Ansermet,* Matthias B. Moor,* Gabriel Centeno,* Muriel Auberson,* † † ‡ Dorothy Zhang Hu, Roland Baron, Svetlana Nikolaeva,* Barbara Haenzi,* | Natalya Katanaeva,* Ivan Gautschi,* Vladimir Katanaev,*§ Samuel Rotman, Robert Koesters,¶ †† Laurent Schild,* Sylvain Pradervand,** Olivier Bonny,* and Dmitri Firsov* BRIEF COMMUNICATION *Department of Pharmacology and Toxicology and **Genomic Technologies Facility, University of Lausanne, Lausanne, Switzerland; †Department of Oral Medicine, Infection, and Immunity, Harvard School of Dental Medicine, Boston, Massachusetts; ‡Institute of Evolutionary Physiology and Biochemistry, St. Petersburg, Russia; §School of Biomedicine, Far Eastern Federal University, Vladivostok, Russia; |Services of Pathology and ††Nephrology, Department of Medicine, University Hospital of Lausanne, Lausanne, Switzerland; and ¶Université Pierre et Marie Curie, Paris, France ABSTRACT Tight control of extracellular and intracellular inorganic phosphate (Pi) levels is crit- leaves.4 Most recently, Legati et al. have ical to most biochemical and physiologic processes. Urinary Pi is freely filtered at the shown an association between genetic kidney glomerulus and is reabsorbed in the renal tubule by the action of the apical polymorphisms in Xpr1 and primary fa- sodium-dependent phosphate transporters, NaPi-IIa/NaPi-IIc/Pit2. However, the milial brain calcification disorder.5 How- molecular identity of the protein(s) participating in the basolateral Pi efflux remains ever, the role of XPR1 in the maintenance unknown. Evidence has suggested that xenotropic and polytropic retroviral recep- of Pi homeostasis remains unknown. Here, tor 1 (XPR1) might be involved in this process. Here, we show that conditional in- we addressed this issue in mice deficient for activation of Xpr1 in the renal tubule in mice resulted in impaired renal Pi Xpr1 in the nephron. -

Normal Rangesа–А
Effective August 2007 5361 NW 33 rd Avenue Fort Lauderdale, FL 33309 954-777-0018 NORMAL RANGES – fax: 954-777-0211 ADULT MALE FEMALE CBC WBC 3.6 – 11.0 K/UL 3.6 – 11.0 RBC 4.5 – 5.9 M/UL 4.5 – 5.9 HEMOGLOBIN 13.0 – 18.0 G/DL 12.0 – 15.6 HEMATOCRIT 40 – 52 % 36.0 – 46.0 MCV 81 – 97 FL 81 93 MCH 26 – 34 PG 26 34 MCHC 31 – 37 gm/dl 31 37 RDW 11.5 – 15 % 11.5 – 15.0 PLATELET CT 150 – 400 K/UL 140 400 PLT VOL 7.4 – 10.4 FL 7.4 – 10.4 NEUTROPHIL % 36 – 66 36 75 LYMPHOCYTE % 23 – 43 27 47 MONOCYTE % 0.0 – 10 0.0 10 EOSINOPHIL % 0 – 5.0 0.0 – 5.0 BASOPHIL % 0 – 1.0 0.0 – 1.0 RETIC 0.4 – 1.9 0.4 – 1.9 COMPREHENSIVE METABOLIC PROFILE /LIVER PROFILE SODIUM 135 – 145 MMOL/L 135 145 POTASSIUM 3.5 – 5.5 MMOL/L 3.5 – 5.5 (OUTREACH) CHLORIDE 95 – 110 MMOL/L 95 110 CO2 19 – 34.MMOL/L 19 34 GLUCOSE 70 – 110 MG/DL 70 110 BUN 6 – 22 MG/DL 6 22 CREATININE 0.6 – 1.3 MG/DL 0.6 – 1.3 MG/DL CALCIUM 8.4 – 10.2 MG/DL 8.4 – 10.2 TOTAL PROTEIN 5.5 – 8.7 G/DL 5.5 – 8.7 ALBUMIN 3.2 – 5.0 g/dl 3.2 – 5.0 BILI TOTAL 0.1 – 1.2 MG/DL 0.1 – 1.2 BILI DIRECT 0.0 – 0.3 MG/DL 0.0 – 0.3 BILI INDIRECT 0.2 – 1.0 MG/DL 0.2 – 1.0 ALKALINE PHOS 20 – 130 U/L 20 130 AST/SGOT 10 – 40 U/L 10 40 ALT/SGPT 7 55 U/L 7 55 AST/ALT RATIO 0.5 – 0.9 RATIO 0.5 – 0.9 AMYLASE 25 – 115 U/L 25 – 115 U/L LIPASE 114 – 286 U/L 114 – 286 U/L CRP 0 – 0.9 MG/DL 0 – 0.9 MG/DL PREALBUMIN 18.0 – 35.7 mg/dL 18.0 – 35.7 mg/dL Revised 807 MAGNESIUM 1.8 – 2.4 mg/dL 1.8 – 2.4 mg/Dl LDH 100 – 200 Units /L 100 – 200 Units/L PHOSPHOROUS 2.3 – 5.0 mg/dL 2.3 – 5.0 mg/dL LACTIC ACID 0.4 -
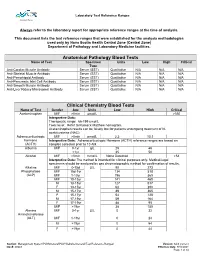
Laboratory Test Reference Ranges
Laboratory Test Reference Ranges Central Zone Always refer to the laboratory report for appropriate reference ranges at the time of analysis. This document lists the test reference ranges that were established for the analysis methodologies used only by Nova Scotia Health Central Zone (Central Zone) Department of Pathology and Laboratory Medicine facilities. Anatomical Pathology Blood Tests Name of Test Specimen Units Low High Critical Type Anti-Cardiac Muscle Antibody Serum (SST) Qualitative N/A N/A N/A Anti-Skeletal Muscle Antibody Serum (SST) Qualitative N/A N/A N/A Anti-Pemphigoid Antibody Serum (SST) Qualitative N/A N/A N/A Anti-Pancreatic Islet Cell Antibody Serum (SST) Qualitative N/A N/A N/A Anti-Smooth Muscle Antibody Serum (SST) Qualitative N/A N/A N/A Anti-Liver Kidney Microsomal Antibody Serum (SST) Qualitative N/A N/A N/A Clinical Chemistry Blood Tests Name of Test Gender Age Units Low High Critical Acetaminophen M/F >0min µmol/L >350 Interpretive Data: Therapeutic range: 66-199 umol/L Toxic level: Refer to Rumack Matthew nomogram. Acetaminophen results can be falsely low for patients undergoing treatment of N- acetylcysteine (NAC). Adrenocorticotropic M/F >0min pmol/L 2.3 10.1 hormone Interpretive Data: Adrenocorticotropic Hormone (ACTH) reference ranges are based on (ACTH) samples collected prior to 10 AM. Albumin M/F 0-1yr g/L 25 46 >1yr 35 50 Alcohol M/F >0min mmol/L None Detected >54 Interpretive Data: The method is intended for clinical purposes only. Medical-legal specimens should be analyzed by gas chromatographic method for confirmation of results. -

Determination of Reference Ranges for Selected Clinical Laboratory Tests for a Medical Laboratory in Namibia Using Pre-Tested Data
DETERMINATION OF REFERENCE RANGES FOR SELECTED CLINICAL LABORATORY TESTS FOR A MEDICAL LABORATORY IN NAMIBIA USING PRE-TESTED DATA by CORNELIA DE WAAL-MILLER Student no: 197084516 Thesis submitted in fulfillment of the requirements of the degree Master of Technology: Biomedical Technology in the Faculty of Health and Wellness Sciences at the Cape Peninsula University of Technology Supervisor: Professor A.J. Esterhuyse Co-Supervisor: Professor B. Noden Bellville March 2015 CPUT copyright information The thesis may not be published either in part (in scholarly, scientific or technical journals), or as a whole (as a monograph), unless permission has been obtained from the University. (i) Declaration I, Cornelia de Waal-Miller, declare that the content of this thesis represents my own unaided work, and that this thesis has not previously been submitted for academic examination towards any qualification. Furthermore, this thesis represents my own opinions and not necessarily those of the Cape Peninsula University of Technology. 12th March 2015 Signed Date 2 | of 84 Pages (ii) Abstract Aim: The aim of the study was to compile pre-tested laboratory results stored in the laboratory database of the Namibia Institute of Pathology (NIP). The study also aimed to assess the usefulness and validity of using retrospective laboratory results of different patients in varying degrees of health and which were produced using various methods in different laboratories in Namibia. Methods: 254,271 test results (female: 134,261, male = 117,091, unknown gender= 2,919) consisting of Haemoglobin, serum Urea, serum Creatinine, plasma Glucose (fasting and random), serum Cholesterol, serum Triglycerides and serum Uric Acid was extracted from NIP Laboratory Information System over a period of four years and of the 13 different regions of Namibia were analyzed. -

Analysis of OAT, OCT, OCTN, and Other Family Members Reveals 8
bioRxiv preprint doi: https://doi.org/10.1101/2019.12.23.887299; this version posted December 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Reclassification of SLC22 Transporters: Analysis of OAT, OCT, OCTN, and other Family Members Reveals 8 Functional Subgroups Darcy Engelhart1, Jeffry C. Granados2, Da Shi3, Milton Saier Jr.4, Michael Baker6, Ruben Abagyan3, Sanjay K. Nigam5,6 1Department of Biology, University of California San Diego, La Jolla 92093 2Department of Bioengineering, University of California San Diego, La Jolla 92093 3School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla 92093 4Department of Molecular Biology, Division of Biological Sciences, University of California at San Diego, San Diego, CA, USA 5Department of Pediatrics, University of California San Diego, La Jolla 92093 6Department of Medicine, University of California San Diego, La Jolla 92093 *To whom correspondence should be addressed: [email protected] Running title: Functional subgroups for SLC22 1 bioRxiv preprint doi: https://doi.org/10.1101/2019.12.23.887299; this version posted December 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Among transporters, the SLC22 family is emerging as a central hub of endogenous physiology. -

Disease-Induced Modulation of Drug Transporters at the Blood–Brain Barrier Level
International Journal of Molecular Sciences Review Disease-Induced Modulation of Drug Transporters at the Blood–Brain Barrier Level Sweilem B. Al Rihani 1 , Lucy I. Darakjian 1, Malavika Deodhar 1 , Pamela Dow 1 , Jacques Turgeon 1,2 and Veronique Michaud 1,2,* 1 Tabula Rasa HealthCare, Precision Pharmacotherapy Research and Development Institute, Orlando, FL 32827, USA; [email protected] (S.B.A.R.); [email protected] (L.I.D.); [email protected] (M.D.); [email protected] (P.D.); [email protected] (J.T.) 2 Faculty of Pharmacy, Université de Montréal, Montreal, QC H3C 3J7, Canada * Correspondence: [email protected]; Tel.: +1-856-938-8697 Abstract: The blood–brain barrier (BBB) is a highly selective and restrictive semipermeable network of cells and blood vessel constituents. All components of the neurovascular unit give to the BBB its crucial and protective function, i.e., to regulate homeostasis in the central nervous system (CNS) by removing substances from the endothelial compartment and supplying the brain with nutrients and other endogenous compounds. Many transporters have been identified that play a role in maintaining BBB integrity and homeostasis. As such, the restrictive nature of the BBB provides an obstacle for drug delivery to the CNS. Nevertheless, according to their physicochemical or pharmacological properties, drugs may reach the CNS by passive diffusion or be subjected to putative influx and/or efflux through BBB membrane transporters, allowing or limiting their distribution to the CNS. Drug transporters functionally expressed on various compartments of the BBB involve numerous proteins from either the ATP-binding cassette (ABC) or the solute carrier (SLC) superfamilies. -

Calcium, Plasma, Total
Division of Laboratory Medicine Biochemistry Calcium, plasma, total Calcium is the most abundant mineral element in the body with about 99 percent in the bones primarily as hydroxyapatite. The remaining calcium is distributed between the various tissues and the extracellular fluids where it performs a vital role for many life sustaining processes. Among the extra skeletal functions of calcium are involvement in blood coagulation, neuromuscular conduction, excitability of skeletal and cardiac muscle, enzyme activation, and the preservation of cell membrane integrity and permeability. Calcium levels and hence the body content are controlled by parathyroid hormone (PTH), calcitonin, and vitamin D. To maintain calcium homeostasis, we require sufficient intake, normal metabolism and appropriate excretion. An imbalance in any of these modulators leads to alterations of the body and circulating calcium levels. Increases in serum PTH or vitamin D are usually associated with hypercalcemia. Increased calcium levels may also be observed in multiple myeloma and other neoplastic diseases. Hypocalcemia may be observed e.g. in hypoparathyroidism, nephrosis, and pancreatitis. Total Calcium is reported as direct and after adjustment for plasma albumin level. Adjusted total calcium has the same reference interval as total calcium and is a surrogate for ionised calcium. General information Collection container: Adults – serum (with gel separator, 4.9mL brown top Sarstedt tube) Paediatrics – lithium heparin plasma (1.2mL orange top Sarstedt tube) Type and volume of sample: The tubes should be thoroughly mixed before transport to the lab. 1mL whole blood is required as a minimum volume. Specimen transport/special precautions: N/A Laboratory information Method principle: Calcium ions react with 5‑nitro‑5’‑methyl‑BAPTA (NM‑BAPTA) under alkaline conditions to form a complex. -

Lab Dept: Chemistry Test Name: VENOUS BLOOD GAS (VBG)
Lab Dept: Chemistry Test Name: VENOUS BLOOD GAS (VBG) General Information Lab Order Codes: VBG Synonyms: Venous blood gas CPT Codes: 82803 - Gases, blood, any combination of pH, pCO2, pO2, CO2, HCO3 (including calculated O2 saturation) Test Includes: VpH (no units), VpCO2 and VpO2 measured in mmHg, VsO2 and VO2AD measured in %, HCO3 and BE measured in mmol/L, Temperature (degrees C) and ST (specimen type) Logistics Test Indications: Useful for evaluating oxygen and carbon dioxide gas exchange; respiratory function, including hypoxia; and acid/base balance. It is also useful in assessment of asthma; chronic obstructive pulmonary disease and other types of lung disease; embolism, including fat embolism; and coronary artery disease. Lab Testing Sections: Chemistry Phone Numbers: MIN Lab: 612-813-6280 STP Lab: 651-220-6550 Test Availability: Daily, 24 hours Turnaround Time: 30 minutes Special Instructions: See Collection and Patient Preparation Specimen Specimen Type: Whole blood Container: Preferred: Sims Portex® syringe (PB151) or Smooth-E syringe (956- 463) Draw Volume: 0.4 mL (Minimum: 0.2 mL) blood Note: Submission of 0.2 mL of blood does not allow for repeat analysis. Processed Volume: 0.2 mL blood per analysis Collection: Avoid using a tourniquet. Anaerobically collect blood into a heparinized blood gas syringe (See Container. Once the puncture has been performed or the line specimen drawn, immediately remove all air from the syringe. Remove the needle, cap tightly and gently mix. Do not expose the specimen to air. Forward the specimen immediately at ambient temperature. Specimens cannot be stored. Note: When drawing from an indwelling catheter, the line must be thoroughly flushed with blood before drawing the sample. -

Morin Improves Urate Excretion and Kidney Function Through Regulation of Renal Organic Ion Transporters in Hyperuricemic Mice
J Pharm Pharmaceut Sci (www.cspsCanada.org) 13(3) 411 - 427, 2010 Morin Improves Urate Excretion and Kidney Function through Regulation of Renal Organic Ion Transporters in Hyperuricemic Mice Cai-Ping Wang, Xing Wang, Xian Zhang, Yun-Wei Shi, Lei Liu, Ling-Dong Kong State Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, Nanjing 210093, P. R. China Received, May 17, 2010; Revised, September 23, 2010; Accepted, October 5, 2010; October 5, 2010. ABSTRACT - Purpose. Morin (2′,3,4′,5,7-pentahydroxyflavone), a plant-derived flavonoid, has beneficial effects on hyperuricemia and renal dysfunction in animals. Since the decreased renal excretion of uric acid is the hallmark of hyperuricemia, here we studied the effects of oral morin administration on renal organic ion transporters in potassium oxonate-induced hyperuricemic mice. Methods. Hyperuricemia in mice was induced by potassium oxonate. Uric acid and creatinine concentrations in urine and serum, and fractional excretion of uric acid (FEUA) were performed to evaluate renal urate handling. Changes in expression levels of renal organic ion transporters were detected by Western blotting and semi-quantitative reverse transcription polymerase chain reaction (RT-PCR) methods. Results. Morin treatment significantly increased urinary uric acid/creatinine ratio and FEUA, resulting in reduction of serum uric acid levels in hyperuricemic mice. And kidney conditions were also improved after morin treatment in this model. Protein and mRNA levels of glucose transporter 9 (mGLUT9) and urate transporter 1 (mURAT1) were significantly decreased, and of organic anion transporter 1 (mOAT1) were remarkably increased in the kidney of morin-treated hyperuricemic mice. Morin treatment also blocked down-regulations of renal organic cation and carnitine transporters (mOCT1, mOCT2, mOCTN1 and mOCTN2) in hyperuricemic mice. -

Uric Acid 7D76-20 30-3928/R4
URIC ACID 7D76-20 30-3928/R4 URIC ACID This package insert contains information to run the Uric Acid assay on the ARCHITECT c Systems™ and the AEROSET System. NOTE: Changes Highlighted NOTE: This package insert must be read carefully prior to product use. Package insert instructions must be followed accordingly. Reliability of assay results cannot be guaranteed if there are any deviations from the instructions in this package insert. Customer Support United States: 1-877-4ABBOTT Canada: 1-800-387-8378 (English speaking customers) 1-800-465-2675 (French speaking customers) International: Call your local Abbott representative Symbols in Product Labeling Calibrators 1 and 2 Catalog number/List number Concentration Serial number Authorized Representative in the Consult instructions for use European Community Ingredients Manufacturer In vitro diagnostic medical device Temperature limitation Batch code/Lot number Use by/Expiration date Reagent 1 ABBOTT LABORATORIES ABBOTT Abbott Park, IL 60064, USA Max-Planck-Ring 2 65205 Wiesbaden Germany +49-6122-580 July 2007 ©2002, 2007 Abbott Laboratories 1 NAME SPECIMEN COLLECTION AND HANDLING URIC ACID Suitable Specimens Serum, plasma, and urine are acceptable specimens. INTENDED USE • Serum: Use serum collected by standard venipuncture techniques The Uric Acid assay is used for the quantitation of uric acid in human into glass or plastic tubes with or without gel barriers. Ensure serum, plasma, or urine. complete clot formation has taken place prior to centrifugation. When processing samples, separate serum from blood cells or SUMMARY AND EXPLANATION OF TEST gel according to the specimen collection tube manufacturer’s Uric acid is a metabolite of purines, nucleic acids, and nucleoproteins.