Modulation of Urate Transport by Drugs
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Viewed Under 23 (B) Or 203 (C) fi M M Male Cko Mice, and Largely Unaffected Magni Cation; Scale Bars, 500 M (B) and 50 M (C)
BRIEF COMMUNICATION www.jasn.org Renal Fanconi Syndrome and Hypophosphatemic Rickets in the Absence of Xenotropic and Polytropic Retroviral Receptor in the Nephron Camille Ansermet,* Matthias B. Moor,* Gabriel Centeno,* Muriel Auberson,* † † ‡ Dorothy Zhang Hu, Roland Baron, Svetlana Nikolaeva,* Barbara Haenzi,* | Natalya Katanaeva,* Ivan Gautschi,* Vladimir Katanaev,*§ Samuel Rotman, Robert Koesters,¶ †† Laurent Schild,* Sylvain Pradervand,** Olivier Bonny,* and Dmitri Firsov* BRIEF COMMUNICATION *Department of Pharmacology and Toxicology and **Genomic Technologies Facility, University of Lausanne, Lausanne, Switzerland; †Department of Oral Medicine, Infection, and Immunity, Harvard School of Dental Medicine, Boston, Massachusetts; ‡Institute of Evolutionary Physiology and Biochemistry, St. Petersburg, Russia; §School of Biomedicine, Far Eastern Federal University, Vladivostok, Russia; |Services of Pathology and ††Nephrology, Department of Medicine, University Hospital of Lausanne, Lausanne, Switzerland; and ¶Université Pierre et Marie Curie, Paris, France ABSTRACT Tight control of extracellular and intracellular inorganic phosphate (Pi) levels is crit- leaves.4 Most recently, Legati et al. have ical to most biochemical and physiologic processes. Urinary Pi is freely filtered at the shown an association between genetic kidney glomerulus and is reabsorbed in the renal tubule by the action of the apical polymorphisms in Xpr1 and primary fa- sodium-dependent phosphate transporters, NaPi-IIa/NaPi-IIc/Pit2. However, the milial brain calcification disorder.5 How- molecular identity of the protein(s) participating in the basolateral Pi efflux remains ever, the role of XPR1 in the maintenance unknown. Evidence has suggested that xenotropic and polytropic retroviral recep- of Pi homeostasis remains unknown. Here, tor 1 (XPR1) might be involved in this process. Here, we show that conditional in- we addressed this issue in mice deficient for activation of Xpr1 in the renal tubule in mice resulted in impaired renal Pi Xpr1 in the nephron. -

Zurampic (Lesinurad)
Market Applicability Market DC GA KY MD NJ NY WA Applicable X X X X X X NA Zurampic (lesinurad) Override(s) Approval Duration Prior Authorization 1 year Quantity Limit Medications Quantity Limit Zurampic (lesinurad) May be subject to quantity limit APPROVAL CRITERIA Requests for Zurampic (lesinurad) may be approved if the following criteria are met: I. Individual has had a previous trial (medication samples/coupons/discount cards are excluded from consideration as a trial) and inadequate response (unable to achieve target serum uric acid levels) to one preferred xanthine oxidase inhibitor (allopurinol, febuxostat*); AND II. Individual has hyperuricemia associated with gout; AND III. Individual is unable to achieve target serum uric acid levels while on a xanthine oxidase inhibitor (such as allopurinol or febuxostat*); AND IV. Individual remains symptomatic (such as, but not limited to joint pain, swelling, limited range of motion, erythema) despite use of xanthine oxidase inhibitor; AND V. Individual will continue use of a xanthine oxidase inhibitor in conjunction with Zurampic. *requires prior authorization Zurampic (lesinurad) may not be approved for the following: I. Individual has severe renal impairment (creatinine clearance less than 30 mL/min), end stage renal disease, is on dialysis or is a kidney transplant recipient; OR II. Individual has Lesch-Nyhan syndrome or tumor lysis syndrome. PAGE 1 of 2 08/14/2019 This policy does not apply to health plans or member categories that do not have pharmacy benefits, nor does it apply to Medicare. Note that market specific restrictions or transition-of-care benefit limitations may apply. CRX-ALL-0429-19 Market Applicability Market DC GA KY MD NJ NY WA Applicable X X X X X X NA Note: Zurampic (lesinurad) has a black box warning indicating risk of acute renal failure is more common when Zurampic is used without a xanthine oxidase inhibitor. -

Identification of a Novel Nucleobase-Ascorbate Transporter
bioRxiv preprint doi: https://doi.org/10.1101/287870; this version posted December 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Identification of a novel Nucleobase-Ascorbate 2 Transporter family member in fish and amphibians 3 Diogo Oliveira*,1, André M. Machado*,1, Tiago Cardoso1, Mónica Lopes-Marques1, L. Filipe 4 C. Castro1,2● and Raquel Ruivo1● 5 1CIIMAR – Interdisciplinary Centre of Marine and Environmental Research, U. Porto – University 6 of Porto, Porto, Portugal 7 2Department of Biology, Faculty of Sciences, U. Porto - University of Porto, Portugal 8 *Equal contribution 9 ●Corresponding authors at: CIIMAR, Terminal de Cruzeiros do Porto de Leixões, Av. General 10 Norton de Matos s/n, 4450-208 Matosinhos, Portugal. Tel.: +351 223 401 831 11 12 Running title: Novel uric acid transporter in fish and amphibians 13 14 15 bioRxiv preprint doi: https://doi.org/10.1101/287870; this version posted December 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 16 Abstract:Nucleobase-Ascorbate Transporter (NAT) family includes ascorbic acid, nucleobases 17 and uric acid transporters: with a broad evolutionary distribution. In vertebrates, four members 18 have been previously recognized, the ascorbate transporters Slc23a1 and Slc3a2, the nucleobase 19 transporter Slc23a4 and an orphan transporter Slc23a3. -

Differential Expression of Hydroxyurea Transporters in Normal and Polycythemia Vera Hematopoietic Stem and Progenitor Cell Subpopulations
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2021 Differential expression of hydroxyurea transporters in normal and polycythemia vera hematopoietic stem and progenitor cell subpopulations Tan, Ge ; Meier-Abt, Fabienne Abstract: Polycythemia vera (PV) is a myeloproliferative neoplasm marked by hyperproliferation of the myeloid lineages and the presence of an activating JAK2 mutation. Hydroxyurea (HU) is a standard treat- ment for high-risk patients with PV. Because disease-driving mechanisms are thought to arise in PV stem cells, effective treatments should target primarily the stem cell compartment. We tested for theantipro- liferative effect of patient treatment with HU in fluorescence-activated cell sorting-isolated hematopoietic stem/multipotent progenitor cells (HSC/MPPs) and more committed erythroid progenitors (common myeloid/megakaryocyte-erythrocyte progenitors [CMP/MEPs]) in PV using RNA-sequencing and gene set enrichment analysis. HU treatment led to significant downregulation of gene sets associated with cell proliferation in PV HSCs/MPPs, but not in PV CMP/MEPs. To explore the mechanism underlying this finding, we assessed for expression of solute carrier membrane transporters, which mediate trans- membrane movement of drugs such as HU into target cells. The active HU uptake transporter OCTN1 was upregulated in HSC/MPPs compared with CMP/MEPs of untreated patients with PV, and the HU diffusion facilitator urea transporter B (UTB) was downregulated in HSC/MPPs compared withCM- P/MEPs in all patient and control groups tested. These findings indicate a higher accumulation ofHU within PV HSC/MPPs compared with PV CMP/MEPs and provide an explanation for the differential effects of HU in HSC/MPPs and CMP/MEPs of patients with PV. -
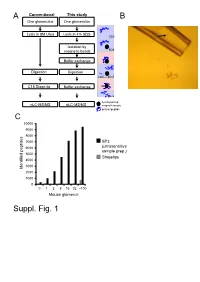
Suppl. Fig. 1 A
A Conventional This study B One glomerulus One glomerulus Lysis in 8M Urea Lysis in 4% SDS SDS Isolation by magnetic beads SDS Buffer exchange Digestion Digestion Try LysC pH>7 C18 Stage tip Buffer exchange pH<4 functionalized nLC-MS/MS nLC-MS/MS magnetic beads protein/peptide C 10000 9000 8000 7000 SP3 6000 (ultrasensitive sample prep.) 5000 Stagetips 4000 3000 Identified peptides 2000 1000 0 0 1 2 8 16 32 ~100 Mouse glomeruli Suppl. Fig. 1 S1. Sample preparation methods. A. Scheme of conventional (C18 Stage Tip) and ultrasensitive proteomic sample preparation methods. B. Manual microdissection and isolation of single glomeruli. The arrow indicates a single isolated glomerulus subjected to proteome analysis. The tip capacity is 10µl. C. Stagetips, a conventional proteomic sample preparation method, is compared with ultrasensitive (SP3) proteome analysis. The indicated amounts of mouse glomeruli were prepared by the respective sample preparation protocols, and peptide numbers after nLC-MS/MS are plotted (all FDR<0.01). A 100 1 Tubule B 80 60 40 Intensity CCD 20 40 0 100 20 S1 TAL 80 0 Tubule proximal tubule Loading... 60 0 Intensity 40 S2 20 -20 control Component 2 (22.5%) S3 0 0 10 20 30 40 50 60 -40 -20 0 20 40 Time (min) Component 2 (47.1%) C D 6000 Mouse proximal tubule 5000 8 4000 3000 7 (iBAQ) Loading... 10 2000 log 6 Number of peptides 1000 0 5 Control mouse mouse mouse human proximal mTAL CCD proximal 0 1500 tubule tubule Rank protein (S1) (S1) R=0.7 E F 8 ACTB Human proximal tubule 8 7 SLC9A3R1 ATP1A1 SLC3A2 SLC25A5 ATP1B1 7 SLC5A2 SLC25A3 CUBN CLTC SLC27A2 SLC5A12 LRP2 6 SLC25A13 SLC25A10 SLC5A1 SLC22A6 6 SLC25A4 Loading.. -

Anti-SLC22A13 (Aa 38-139) Polyclonal Antibody (DPAB-DC3801) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Anti-SLC22A13 (aa 38-139) polyclonal antibody (DPAB-DC3801) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Antigen Description This gene encodes a member of the organic-cation transporter family. It is located in a gene cluster with another member of the family, organic cation transporter like 4. The encoded protein is a transmembrane protein involved in the transport of small molecules. This protein can function to mediate urate uptake and is a high affinity nicotinate exchanger in the kidneys and the intestine. Immunogen SLC22A13 (NP_004247, 38 a.a. ~ 139 a.a) partial recombinant protein with GST tag. The sequence is AHVFMVLDEPHHCAVAWVKNHTFNLSAAEQLVLSVPLDTAGHPEPCLMFRPPPANASLQDILSH RFNETQPCDMGWEYPENRLPSLKNEFNLVCDRKHLKDT Source/Host Mouse Species Reactivity Human Conjugate Unconjugated Applications WB (Recombinant protein), ELISA, Size 50 μl Buffer 50 % glycerol Preservative None Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. GENE INFORMATION Gene Name SLC22A13 solute carrier family 22 (organic anion/urate transporter), member 13 [ Homo sapiens (human) ] Official Symbol SLC22A13 Synonyms SLC22A13; solute carrier family 22 (organic anion/urate transporter), member 13; OAT10; 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved OCTL1; OCTL3; ORCTL3; ORCTL-3; solute carrier family 22 member 13; organic cation transporter-like 3; organic-cation transporter like 3; organic cationic transporter-like 3; solute carrier family 22, member 13; solute carrier family 22 (organic anion transporter), member 13; Entrez Gene ID 9390 Protein Refseq NP_004247 UniProt ID Q9Y226 Chromosome Location 3p21.3 Function nicotinate transporter activity; organic cation transmembrane transporter activity; 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 2 © Creative Diagnostics All Rights Reserved. -

New Therapeutic Agents Marketed in 2015: Part 4 Amy M
CPE New therapeutic agents marketed in 2015: Part 4 Amy M. Lugo and Brian Park Amy M. Lugo, PharmD, BCPS, BC-ADM, Abstract FAPhA, Clinical Pharmacy Specialist, Pharmacy Operations Division, Formu- lary Management Branch, Defense Health Objective: To provide information about the most important properties of new Agency, San Antonio therapeutic agents approved by FDA and first marketed in 2015. Brian Park, PharmD candidate, University of Data sources: Product labeling supplemented selectively with published stud- New England College of Pharmacy, Portland, ies and drug information reference sources. ME Data synthesis: This review covers 11 new therapeutic agents approved by FDA Correspondence: Amy M. Lugo, 7800 and first marketed in the United States in 2015: selexipag, cobimetinib, osimertinib, IH-10 W, Ste. 335, San Antonio, TX 78230; necitumumab, alectinib, insulin degludec, lesinurad, mepolizumab, brexpiprazole, [email protected] cariprazine, and flibanserin. Indications and information on dosage and adminis- Disclosure: The authors declare no conflicts tration for these agents are reviewed, as well as efficacy, safety, the most important of interest or financial interests in any product or service mentioned in this article. pharmacokinetic properties, drug interactions, and other precautions. Practical considerations for use of these new agents also are discussed. Whenever possible, Department of Defense (DoD) disclaimer: The information discussed here represents properties of the new drugs are compared with those of older agents marketed for the views of the author and does not neces- the same indications. sarily reflect the views of DoD or the Depart- Summary: Selexipag is the second oral prostacyclin agonist for the treatment ments of the Army, Navy, or Air Force. -

Expression of SLC2A9 Isoforms in the Kidney and Their Localization in Polarized Epithelial Cells
Expression of SLC2A9 Isoforms in the Kidney and Their Localization in Polarized Epithelial Cells Toru Kimura1, Michi Takahashi1, Kunimasa Yan2, Hiroyuki Sakurai1* 1 Department of Pharmacology and Toxicology, Kyorin University School of Medicine, Mitaka, Tokyo, Japan, 2 Department of Pediatrics, Kyorin University School of Medicine, Mitaka, Tokyo, Japan Abstract Background: Many genome-wide association studies pointed out that SLC2A9 gene, which encodes a voltage-driven urate transporter, SLC2A9/GLUT9 (a.k.a. URATv1), as one of the most influential genes for serum urate levels. SLC2A9 is reported to encode two splice variants: SLC2A9-S (512 amino acids) and SLC2A9-L (540 amino acids), only difference being at their N- termini. We investigated isoform-specific localization of SLC2A9 in the human kidney and role of N-terminal amino acids in differential sorting in vitro. Methodology/Principal Findings: Isoform specific antibodies against SLC2A9 were developed and human kidney sections were stained. SLC2A9-S was expressed in the apical side of the collecting duct while SLC2A9-L was expressed in the basolateral side of the proximal tubule. GFP fused SLC2A9s were expressed in MDCK cells and intracellular localization was observed. SLC2A9-S was expressed at both apical and basolateral membranes, whereas SLC2A9-L was expressed only at the basolateral membrane. Although SLC2A9-L has a putative di-leucine motif at 33th and 34th leucine, deletion of the motif or replacement of leucine did not affect its subcellular localization. When up to 16 amino acids were removed from the N- terminal of SLC2A9-S or when up to 25 amino acids were removed from the N-terminal of SLC2A9-L, there was no change in their sorting. -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

2.1 Drosophila Melanogaster
Overend, Gayle (2010) Drosophila as a model for the Anopheles Malpighian tubule. PhD thesis, University of Glasgow. http://theses.gla.ac.uk/1604/ Copyright and moral rights for this thesis are retained by the author A copy can be downloaded for personal non-commercial research or study, without prior permission or charge This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the Author The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the Author When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given Glasgow Theses Service http://theses.gla.ac.uk/ [email protected] Drosophila as a model for the Anopheles Malpighian tubule A thesis submitted for the degree of Doctor of Philosophy at the University of Glasgow Gayle Overend Integrative and Systems Biology Faculty of Biomedical and Life Sciences University of Glasgow Glasgow G11 6NU UK August 2009 2 The research reported within this thesis is my own work except where otherwise stated, and has not been submitted for any other degree. Gayle Overend 3 Abstract The insect Malpighian tubule is involved in osmoregulation, detoxification and immune function, physiological processes which are essential for insect development and survival. As the Malpighian tubules contain many ion channels and transporters, they could be an effective tissue for targeting with novel pesticides to control populations of Diptera. Many of the insecticide compounds used to control insect pest species are no longer suited to their task, and so new means of control must be found. -

Stems for Nonproprietary Drug Names
USAN STEM LIST STEM DEFINITION EXAMPLES -abine (see -arabine, -citabine) -ac anti-inflammatory agents (acetic acid derivatives) bromfenac dexpemedolac -acetam (see -racetam) -adol or analgesics (mixed opiate receptor agonists/ tazadolene -adol- antagonists) spiradolene levonantradol -adox antibacterials (quinoline dioxide derivatives) carbadox -afenone antiarrhythmics (propafenone derivatives) alprafenone diprafenonex -afil PDE5 inhibitors tadalafil -aj- antiarrhythmics (ajmaline derivatives) lorajmine -aldrate antacid aluminum salts magaldrate -algron alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol combined alpha and beta blockers labetalol medroxalol -amidis antimyloidotics tafamidis -amivir (see -vir) -ampa ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) subgroup: -ampanel antagonists becampanel -ampator modulators forampator -anib angiogenesis inhibitors pegaptanib cediranib 1 subgroup: -siranib siRNA bevasiranib -andr- androgens nandrolone -anserin serotonin 5-HT2 receptor antagonists altanserin tropanserin adatanserin -antel anthelmintics (undefined group) carbantel subgroup: -quantel 2-deoxoparaherquamide A derivatives derquantel -antrone antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine antineoplastics (arabinofuranosyl derivatives) fazarabine fludarabine aril-, -aril, -aril- antiviral (arildone derivatives) pleconaril arildone fosarilate -arit antirheumatics (lobenzarit type) lobenzarit clobuzarit -arol anticoagulants (dicumarol type) dicumarol -

Distribution of Glucose Transporters in Renal Diseases Leszek Szablewski
Szablewski Journal of Biomedical Science (2017) 24:64 DOI 10.1186/s12929-017-0371-7 REVIEW Open Access Distribution of glucose transporters in renal diseases Leszek Szablewski Abstract Kidneys play an important role in glucose homeostasis. Renal gluconeogenesis prevents hypoglycemia by releasing glucose into the blood stream. Glucose homeostasis is also due, in part, to reabsorption and excretion of hexose in the kidney. Lipid bilayer of plasma membrane is impermeable for glucose, which is hydrophilic and soluble in water. Therefore, transport of glucose across the plasma membrane depends on carrier proteins expressed in the plasma membrane. In humans, there are three families of glucose transporters: GLUT proteins, sodium-dependent glucose transporters (SGLTs) and SWEET. In kidney, only GLUTs and SGLTs protein are expressed. Mutations within genes that code these proteins lead to different renal disorders and diseases. However, diseases, not only renal, such as diabetes, may damage expression and function of renal glucose transporters. Keywords: Kidney, GLUT proteins, SGLT proteins, Diabetes, Familial renal glucosuria, Fanconi-Bickel syndrome, Renal cancers Background Because glucose is hydrophilic and soluble in water, lipid Maintenance of glucose homeostasis prevents pathological bilayer of plasma membrane is impermeable for it. There- consequences due to prolonged hyperglycemia or fore, transport of glucose into cells depends on carrier pro- hypoglycemia. Hyperglycemia leads to a high risk of vascu- teins that are present in the plasma membrane. In humans, lar complications, nephropathy, neuropathy and retinop- there are three families of glucose transporters: GLUT pro- athy. Hypoglycemia may damage the central nervous teins, encoded by SLC2 genes; sodium-dependent glucose system and lead to a higher risk of death.