Impact of Transposition on the Generation of Genetic Variability in Prunus Crop Species
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pru Nus Contains Many Species and Cultivars, Pru Nus Including Both Fruits and Woody Ornamentals
;J. N l\J d.000 A~ :J-6 '. AGRICULTURAL EXTENSION SERVICE UNIVERSITY OF MINNESOTA • The genus Pru nus contains many species and cultivars, Pru nus including both fruits and woody ornamentals. The arboretum's Prunus maacki (Amur Cherry). This small tree has bright, emphasis is on the ornamental plants. brownish-yellow bark that flakes off in papery strips. It is par Prunus americana (American Plum). This small tree furnishes ticularly attractive in winter when the stems contrast with the fruits prized for making preserves and is also an ornamental. snow. The flowers and fruits are produced in drooping racemes In early May, the trees are covered with a "snowball" bloom similar to those of our native chokecherry. This plant is ex of white flowers. If these blooms escape the spring frosts, tremely hardy and well worth growing. there will be a crop of colorful fruits in the fall. The trees Prunus maritima (Beach Plum). This species is native to the sucker freely, and unless controlled, a thicket results. The A coastal plains from Maine to Virginia. It's a sprawling shrub merican Plum is excellent for conservation purposes, and the reaching a height of about 6 feet. It blooms early with small thickets are favorite refuges for birds and wildlife. white flowers. Our plants have shown varying degrees of die Prunus amygdalus (Almond). Several cultivars of almonds back and have been removed for this reason. including 'Halls' and 'Princess'-have been tested. Although Prunus 'Minnesota Purple.' This cultivar was named by the the plants survived and even flowered, each winter's dieback University of Minnesota in 1920. -

Conservation Plant Release Brochure for Catskill Dwarf Sand Cherry
A Conservation Plant Released by the Natural Resources Conservation Service Big Flats Plant Materials Center, Big Flats, New York Conservation Uses ‘Catskill’ Catskill is mainly used in shoreline and streambank stabilization practices and riparian buffer plantings, where Dwarf Sand Cherry low vegetation is preferred. Its growth habit makes it adapted to areas with ice floe problems. Prunus pumila var. depressa L. Due to its prostrate growth, it may become shaded out over time by taller vegetation. The fruits produced by Catskill are valuable for wildlife and is a very attractive plant and is used in ornamental landscaping. Area of Adaptation and Use Catskill grows well on gravelly or sandy soils along streams but has performed well on silt loam and calcareous soils. It will tolerate periodic flooding only for a short period. Its massive root system allows it to tolerate drought conditions. It is found from Ontario, Canada to the New York- Pennsylvania border and is adapted to USDA hardiness zones 3b to 6b. ‘Catskill' dwarf sand cherry planted among rip rap, in New York. ‘Catskill’ dwarf sand cherry (Prunus pumila var. depressa Establishment and Management for Conservation (L.)) was released in 1997, by the USDA Natural Plantings Resources Conservation Service (NRCS), Big Flats Plant Establishment: Planting 1-0 nursery bare root stock of Materials Center and the Pennsylvania Game Commission Catskill is the preferred method of establishment. for its prostrate growth habit and immense root system. Planting should be in the spring, prior to June 1st, or in the fall after October 10th. The stock should be dormant at Description the time of planting. -

Wild Plums Cherry Plums Sand Cherries
wild plums cherry plums sand cherries Wild plums, sand cherries, and cherry plums have ripening. Fruit varies from 1 to 1 ½ inch in diameter, the distinct plum characteristics of sour skins, sweet and fruits with soft, sweet flesh tend to be larger. flesh that sticks tightly to the pit, and pits that are flatter than they are round. Plants vary from shrubs to small trees. Wild plums (Prunus americana) are native to much of the U.S. North America is host to a variety of distinct plum species, but the only species widely planted in Minnesota is P. americana. Wild plums are typically large shrubs or small trees that sucker profusely, often sending out root suckers that sprout 20 or more feet from the mother plant. The fruit flesh is yellow, while the skin color varies from yellow to red, with the most common color being a red blush. Like most wild plants that are propagated from seeds, fruit quality varies tremendously. The best quality fruit has thick, sour skins, with sweet flesh that clings tightly to the pit. Some trees produce fruit with the texture of a golf ball that does not soften during Figure 41. Edible wild plums perennial fruit for northern climates 83 Sand cherries are small, native shrubs which Cherry plum is kind of a catch-all term for a number produce a small fruit that is closer to plums than of small fruited plums that belong to several related cherries. In some literature, all sand cherries are put species. Most produce red or purple fruit. -
Botanical Name Common Name
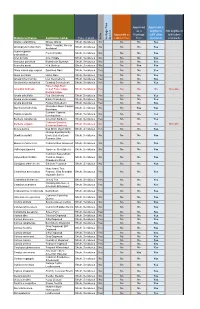
Approved Approved & as a eligible to Not eligible to Approved as Frontage fulfill other fulfill other Type of plant a Street Tree Tree standards standards Heritage Tree Tree Heritage Species Botanical Name Common name Native Abelia x grandiflora Glossy Abelia Shrub, Deciduous No No No Yes White Forsytha; Korean Abeliophyllum distichum Shrub, Deciduous No No No Yes Abelialeaf Acanthropanax Fiveleaf Aralia Shrub, Deciduous No No No Yes sieboldianus Acer ginnala Amur Maple Shrub, Deciduous No No No Yes Aesculus parviflora Bottlebrush Buckeye Shrub, Deciduous No No No Yes Aesculus pavia Red Buckeye Shrub, Deciduous No No Yes Yes Alnus incana ssp. rugosa Speckled Alder Shrub, Deciduous Yes No No Yes Alnus serrulata Hazel Alder Shrub, Deciduous Yes No No Yes Amelanchier humilis Low Serviceberry Shrub, Deciduous Yes No No Yes Amelanchier stolonifera Running Serviceberry Shrub, Deciduous Yes No No Yes False Indigo Bush; Amorpha fruticosa Desert False Indigo; Shrub, Deciduous Yes No No No Not eligible Bastard Indigo Aronia arbutifolia Red Chokeberry Shrub, Deciduous Yes No No Yes Aronia melanocarpa Black Chokeberry Shrub, Deciduous Yes No No Yes Aronia prunifolia Purple Chokeberry Shrub, Deciduous Yes No No Yes Groundsel-Bush; Eastern Baccharis halimifolia Shrub, Deciduous No No Yes Yes Baccharis Summer Cypress; Bassia scoparia Shrub, Deciduous No No No Yes Burning-Bush Berberis canadensis American Barberry Shrub, Deciduous Yes No No Yes Common Barberry; Berberis vulgaris Shrub, Deciduous No No No No Not eligible European Barberry Betula pumila -

Native Pollinator Plants by Season of Bloom
Native Pollinator Plants by Season of Bloom Extended list of forage and host plants for bees, butterflies and moths Very early spring SHRUBS PERENNIALS American hazelnut, Corylus americana, Bloodroot, Sanguinaria Canadensis C. cornuta Sand/moss phlox, Phlox bifida & P. subulata American honeysuckle, Lonicera canadensis Pussy willow, Salix discolor Shadbush, Amerlanchier canadensis, A. laevis Bloodroot, © Lisa Looke Early spring SHRUBS PERENNIALS Bayberry, Morella caroliniensis Blue cohosh, Caulophyllum thalictroides Flowering big-bracted dogwood, Benthamidia Dutchman’s breeches, Dicentra cucullaria florida Crested Iris, Iris cristata* Hobblebush, Viburnum lanatanoides Golden groundsel, Packera aurea Red eldeberry, Sambucus pubens Spicebush, Lindera benzoin Marsh marigold, Caltha palustrus Sweet fern, Comptonia peregrina Pussytoes, Antennaria spp. Sweetgale, Myrica gale Rue anemone, Thalictrum thalictroides Wild plums Violets, Viola adunca, V. cuccularia Beach plum, Prunus maritima Virginia bluebells, Mertensia virginica* Canada plum, Prunus nigra Marsh marigold, © Lisa Looke Sand plum, Prunus pumila Mid-spring SHRUBS PERENNIALS (continued) Bearberry, Arctostaphylos uva-ursi Canada wild ginger, Asarum canadense Black huckleberry, Gaylussacia baccata Common golden Alexanders, Zizia aurea Blueberry, Vaccinium spp. Early meadow-rue, Thalictrum dioicum Eastern shooting star, Dodecatheon meadia* Chokeberry, Aronia arbutifolia & Aronia Foam flower, Tiarella cordifolia melanocarpa Heart-leaved golden Alexanders, Zizia aptera Common snowberry, Symphoricarpos albus Jacob’s ladder, Polemonium reptans* Fragrant sumac, Rhus aromatica* King Solomon’s-seal, Polygonatum biflorum Mountain maple, Acer spicatum var. commutaturn Nannyberry, Viburnum lentago Large-leaved pussytoes, Antennaria Red buckeye, Aesculus pavia* plantaginifolia Nodding onion, Alium cernuum* Spotted crane’s-bill, © Lisa Looke Redbud, Cersis canadensis* Striped maple, Acer pennsylvanicum Red baneberry, Actaea rubra Red columbine, Aquilegia canadensis Solomon’s plume, Maianthemum racemosum PERENNIALS (syn. -

New Varieties
NEW2021 VARIETIES AUTUMN INFERNO® COTONEASTER Cotoneaster ‘Bronfire’ PP30,493 USDA Hardiness Zone: 5 AHS Heat Zone: 7 Height: 5-6’ Spread: 4-5’ Exposure: Full Sun to Part Shade Shape: Upright- Arching Discovered at Bron & Sons Nursery in British Columbia, this new Cotoneaster is a natural cross between C. lucidus and C. apiculatus. It has many of the attributes of C. lucidus that we so love for a hedge - great form, easily pruned, great foliage all season long plus great fall color. But what we really love about Autumn Inferno® is the small red berries formed along the stems in fall. The berries stay on the plant until the birds come and take them, no mess involved. Perfect as a pruned formal hedge or let it go natural. Just enough height to block off your annoying neighbors. FIRSTEDITIONSPLANTS.COM Plant patent has been applied for and trademark has been declared. Propagation and/or use of trademark without license is prohibited. GALACTIC PINK® CHASTETREE Vitex agnus-castus ‘Bailtextwo’ PP30,852 USDA Hardiness Zone: 6 AHS Heat Zone: 9 Height: 6-8’ Spread: 6-8’ Exposure: Full Sun Shape: Upright - Rounded Galactic Pink® has flowers that are a beautiful shade of pink and intermediate in size. The fragrant flowers, born in longGalactic panicles, Pink® bloomhas flowers from Junethat areto Octobera beautiful and shade attract of pollinatorspink and intermediate by the dozens. in size. The The darkfragrant green flowers, foliage is cleanborn and in the long branching panicles, bloomis dense from and June full. toMaturing October atand a slightly attract pollinatorssmaller size by than the dozens.Delta Blues™. -

Shrubs of the Chicago Region
A Selection of Native Shrubs and Noteworthy Non-Native Shrubs 1 WEB VERSION Shrubs of the Chicago Region Volunteer Stewardship Network – Chicago Wilderness Photos by: © Paul Rothrock (Taylor University, IN), © John & Jane Balaban ([email protected]; North Branch Restoration Project), © Kenneth Dritz, © Sue Auerbach, © Melanie Gunn, © Sharon Shattuck, and © William Burger (Field Museum). Produced by: Jennie Kluse © vPlants.org and Sharon Shattuck, with assistance from Ken Klick (Lake County Forest Preserve), Paul Rothrock, Sue Auerbach, John & Jane Balaban, and Laurel Ross. © Environment, Culture and Conservation, The Field Museum, Chicago, IL 60605 USA. [http://www.fmnh.org/temperateguides/]. Chicago Wilderness Guide #5 version 1 (06/2008) EVERGREEN SHRUBS: GROUP 1. LEAVES ARE NEEDLES or SCALES. 1 Juniperus horizontalis TRAILING JUNIPER: 2 Juniperus communis COMMON JUNIPER: Plants have a creeping habit; some leaves are needles but most are Erect shrub or tree (up to 3 m tall); needles whorled on stem; scales with a whitish coat; fruit a bluish-whitish berry-like cone; fruit a bluish or black berry-like cone; grows only in dunes/bluffs male cones on separate plants; grows in sandy soils. bordering Lake Michigan. DECIDUOUS SHRUBS: GROUP 2. LEAVES COMPOUND (more than one leaflet per stalk). STEMS ARMED. 3 Rosa setigera ILLINOIS ROSE: 4 Rosa palustris SWAMP ROSE: Mature plant with long-arching stems; sparse prickles; leaflets Upright shrub; stems very thorny; leaflets 5-7; sepals fall from usually 3, but sometimes 5; styles (female pollen tube) fused into mature fruit; fruit smooth, red berry-like hips; grows in wet and a column; stipules narrow to tip. open ditches, bogs, and swamps. -

Common and Scientific Names of Ornamental Crops
This is a section from the 2012 DISEASE CONTROL RECOMMENDATIONS FOR ORNAMENTAL CROPS Publication E036 The full manual, containing recommendations specific to New Jersey, can be found on the Rutgers NJAES website in the publications section: njaes.rutgers.edu/pubs/publication.asp?pid=E036 Note: The label is a legally-binding contract between the user and the manufacturer. The user must fol- low all rates and restrictions as per label directions. The use of any pesticide inconsistent with the label directions is a violation of Federal law. Mention or display of a trademark, proprietary product, or firm in text or figures does not constitute an endorsement by Rutgers Cooperative Extension and does not imply approval to the exclusion of other suitable products or firms. © 2012 Rutgers, The State University of New Jersey. All rights reserved. Revised: June 2012 Cooperating Agencies: Rutgers, The State University of New Jersey, U.S. Department of Agriculture, and County Boards of Chosen Freeholders, Rutgers Cooperative Exten- sion, a unit of the New Jersey Agricultural Experiment Station, is an equal opportunity program provider and employer. 2012 DISEASE CONTROL RECOMMENDATIONS FOR ORNAMENTAL CROPS Section III Common and Scientifi c Names of Ornamental Crops Note: Host plants are listed on pesticide labels by common or scientifi c name. Below is a list of plants included on the label of one or more pesticide products. Hosts denoted by an “x” are included in Section I, Disease Recommendations by Crop. Common name Other common names Scientifi c name x Abelia Abelia spp. Abutilon Velvetleaf, Chinese Lantern, Mallow, Abutilon spp. Flowering Maple Acacia Thorntree Acacia spp. -

Chapter 21. Trees, Shrubs and Woody Vines
TREES, SHRUBS AND WOODY VINES James B. Calkins Introduction have broad leaves, flowers, variable fruit, and many, Trees, shrubs and woody vines represent the woody but not all, are deciduous or lose their leaves during a members of the plant world. The terms “tree”, “shrub” dormant period, usually during winter. Most and “vine” are non-scientific general descriptive words gymnosperms have narrow leaves called needles, are with no well-defined specific meaning. The American cone-bearing, and are evergreen. Again, there are Standards for Nursery Stock, which is included with notable exceptions to these generalizations. The term this manual, gives some definition to the terms, but in “woody” means that the cells of the stems and general, a “tree” is understood to be a woody plant branches contain ‘lignin’ which serves as a support approximately six feet to over 100 feet in height at structure and gives a permanence to branches even maturity with most major branches derived from a when the plant dies, whereas “herbaceous” plants single erect stem. Variations can include a two to five contain little or no lignin and collapse as soon as the stem clump or a multi-stem form of these same plant dies or goes dormant. species. Shrubs can be defined as almost always having branches deriving from multiple stems and Knowledge of a plant’s classification, characteristics, generally under 15 to 20 feet in height. Vines are and history or native habitat is essential to its highly apically dominant plants, generally with few production in the nursery and to its proper utilization in side branches. -

The Greater Yellowstone Coordinating Committee
THE GREATER YELLOWSTONE COORDINATING COMMITTEE SusTAINABLE OPERATIONS SUBCOmmITTEE REPRESENTATIVES: Jane Ruchman—Gallatin National Forest Landscape Architect & Developed Recreation Program Manager Kaye Suzuki—Beaverhead-Deerlodge National Forest Range Management Specialist In Partnership with: MONTANA STATE UNIVERSITY DEPARTMENT OF PLANT SCIENCES AND PLANT PATHOLOGY Professor Tracy A.O. Dougher, Student Assistant Allen Steckmest, and students Beartooth Mountains Photo courtesy of Bob Kurzenhauser Bozeman, MT Photo courtesy of Blanchford Landscaping 1 CONTENTS Water: Our Most Valuable Resource ............................................................................................... 3 Xeriscaping: A Practical Alternative to Traditional High Water Use Lawns and Landscaping ................ 4 Planning & Design: Zoning for Site Uses, Conditions, and Plant Needs ............................................5 Soil Secrets for Success: Good Soil Supports Healthy Plants With More Efficient Use of aterW ...........7 Plant Selection: Function and Aesthetics ..............................................................................................................9 Right Plant in the Right Place .....................................................................................................10 Greater Yellowstone Area-USDA Plant Hardiness Zones ................................................................11 Adaptations for Water Efficiency ...............................................................................................12 -

Vascular Plant Species of the Comanche National Grassland in United States Department Southeastern Colorado of Agriculture
Vascular Plant Species of the Comanche National Grassland in United States Department Southeastern Colorado of Agriculture Forest Service Donald L. Hazlett Rocky Mountain Research Station General Technical Report RMRS-GTR-130 June 2004 Hazlett, Donald L. 2004. Vascular plant species of the Comanche National Grassland in southeast- ern Colorado. Gen. Tech. Rep. RMRS-GTR-130. Fort Collins, CO: U.S. Department of Agriculture, Forest Service, Rocky Mountain Research Station. 36 p. Abstract This checklist has 785 species and 801 taxa (for taxa, the varieties and subspecies are included in the count) in 90 plant families. The most common plant families are the grasses (Poaceae) and the sunflower family (Asteraceae). Of this total, 513 taxa are definitely known to occur on the Comanche National Grassland. The remaining 288 taxa occur in nearby areas of southeastern Colorado and may be discovered on the Comanche National Grassland. The Author Dr. Donald L. Hazlett has worked as an ecologist, botanist, ethnobotanist, and teacher in Latin America and in Colorado. He has specialized in the flora of the eastern plains since 1985. His many years in Latin America prompted him to include Spanish common names in this report, names that are seldom reported in floristic pub- lications. He is also compiling plant folklore stories for Great Plains plants. Since Don is a native of Otero county, this project was of special interest. All Photos by the Author Cover: Purgatoire Canyon, Comanche National Grassland You may order additional copies of this publication by sending your mailing information in label form through one of the following media. -

Download Article
Advances in Biological Sciences Research, volume 7 1st International Symposium Innovations in Life Sciences (ISILS 2019) The Role Of Inter-Species Hybridization In Expanding The Sortiment Of Plum Julia Burmenko Vladimir Simonov Valery Vysotsky Department of genetics and breeding of Department of genetics and breeding of Department of genetics and breeding of fruit and berry crops fruit and berry crops fruit and berry crops All-Russian Horticultural Institute for All-Russian Horticultural Institute for All-Russian Horticultural Institute for Breeding, Agrotechnology and Nursery Breeding, Agrotechnology and Nursery Breeding, Agrotechnology and Nursery Moscow, Russia Moscow, Russia Moscow, Russia [email protected] [email protected] [email protected] Abstract—As a result of hybridization of species: Prunus In the selection of P. domestica for winter hardiness and domestica L., Prunus salicina Lindl., Prunus rossica Erem., overgrownness, P. spinosa L. is used [5]. In the first Prunus brigantina Vill., Prunus persica (L.) Batsch., Prunus generation, hybrids have 40 chromosomes and low armeniaca Lin., Prunus pumila L., Prunus spinosa L. crossed fecundity. Upon their repeated crossing with plum, the home between Interspecific hybrids combining genomes of 2 or more part of F2 hybrids have 48 chromosomes and normal fertility. species ((Prunus rossica Erem. × Prunus domestica L.) × The fruits of hybrids are close in size and quality to the ((Prunus pumila L. × Prunus salicina Lindl.) × Prunus spinosa varieties of P. domestica and have high winter hardiness. L.), Prunus domestica L. × (Prunus brigantinaVill. × Prunus persica (L.) Batsch.)). Depending on the combination groups, The United States, in particular Zaiger'sInc Genetics, is without using the in vitro method of introducing isolated the undisputed world leader in the achievements of hybrid embryos into the culture, the percentage of surviving interspecific plum hybridization.