Dynamics of Self-Organized and Self-Assembled Structures
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Ordered Distribution of Natural Numbers on the Square Root Spiral
The Ordered Distribution of Natural Numbers on the Square Root Spiral - Harry K. Hahn - Ludwig-Erhard-Str. 10 D-76275 Et Germanytlingen, Germany ------------------------------ mathematical analysis by - Kay Schoenberger - Humboldt-University Berlin ----------------------------- 20. June 2007 Abstract : Natural numbers divisible by the same prime factor lie on defined spiral graphs which are running through the “Square Root Spiral“ ( also named as “Spiral of Theodorus” or “Wurzel Spirale“ or “Einstein Spiral” ). Prime Numbers also clearly accumulate on such spiral graphs. And the square numbers 4, 9, 16, 25, 36 … form a highly three-symmetrical system of three spiral graphs, which divide the square-root-spiral into three equal areas. A mathematical analysis shows that these spiral graphs are defined by quadratic polynomials. The Square Root Spiral is a geometrical structure which is based on the three basic constants: 1, sqrt2 and π (pi) , and the continuous application of the Pythagorean Theorem of the right angled triangle. Fibonacci number sequences also play a part in the structure of the Square Root Spiral. Fibonacci Numbers divide the Square Root Spiral into areas and angle sectors with constant proportions. These proportions are linked to the “golden mean” ( golden section ), which behaves as a self-avoiding-walk- constant in the lattice-like structure of the square root spiral. Contents of the general section Page 1 Introduction to the Square Root Spiral 2 2 Mathematical description of the Square Root Spiral 4 3 The distribution -

Some Curves and the Lengths of Their Arcs Amelia Carolina Sparavigna
Some Curves and the Lengths of their Arcs Amelia Carolina Sparavigna To cite this version: Amelia Carolina Sparavigna. Some Curves and the Lengths of their Arcs. 2021. hal-03236909 HAL Id: hal-03236909 https://hal.archives-ouvertes.fr/hal-03236909 Preprint submitted on 26 May 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Some Curves and the Lengths of their Arcs Amelia Carolina Sparavigna Department of Applied Science and Technology Politecnico di Torino Here we consider some problems from the Finkel's solution book, concerning the length of curves. The curves are Cissoid of Diocles, Conchoid of Nicomedes, Lemniscate of Bernoulli, Versiera of Agnesi, Limaçon, Quadratrix, Spiral of Archimedes, Reciprocal or Hyperbolic spiral, the Lituus, Logarithmic spiral, Curve of Pursuit, a curve on the cone and the Loxodrome. The Versiera will be discussed in detail and the link of its name to the Versine function. Torino, 2 May 2021, DOI: 10.5281/zenodo.4732881 Here we consider some of the problems propose in the Finkel's solution book, having the full title: A mathematical solution book containing systematic solutions of many of the most difficult problems, Taken from the Leading Authors on Arithmetic and Algebra, Many Problems and Solutions from Geometry, Trigonometry and Calculus, Many Problems and Solutions from the Leading Mathematical Journals of the United States, and Many Original Problems and Solutions. -

Archimedean Spirals ∗
Archimedean Spirals ∗ An Archimedean Spiral is a curve defined by a polar equation of the form r = θa, with special names being given for certain values of a. For example if a = 1, so r = θ, then it is called Archimedes’ Spiral. Archimede’s Spiral For a = −1, so r = 1/θ, we get the reciprocal (or hyperbolic) spiral. Reciprocal Spiral ∗This file is from the 3D-XploreMath project. You can find it on the web by searching the name. 1 √ The case a = 1/2, so r = θ, is called the Fermat (or hyperbolic) spiral. Fermat’s Spiral √ While a = −1/2, or r = 1/ θ, it is called the Lituus. Lituus In 3D-XplorMath, you can change the parameter a by going to the menu Settings → Set Parameters, and change the value of aa. You can see an animation of Archimedean spirals where the exponent a varies gradually, from the menu Animate → Morph. 2 The reason that the parabolic spiral and the hyperbolic spiral are so named is that their equations in polar coordinates, rθ = 1 and r2 = θ, respectively resembles the equations for a hyperbola (xy = 1) and parabola (x2 = y) in rectangular coordinates. The hyperbolic spiral is also called reciprocal spiral because it is the inverse curve of Archimedes’ spiral, with inversion center at the origin. The inversion curve of any Archimedean spirals with respect to a circle as center is another Archimedean spiral, scaled by the square of the radius of the circle. This is easily seen as follows. If a point P in the plane has polar coordinates (r, θ), then under inversion in the circle of radius b centered at the origin, it gets mapped to the point P 0 with polar coordinates (b2/r, θ), so that points having polar coordinates (ta, θ) are mapped to points having polar coordinates (b2t−a, θ). -

Analysis of Spiral Curves in Traditional Cultures
Forum _______________________________________________________________ Forma, 22, 133–139, 2007 Analysis of Spiral Curves in Traditional Cultures Ryuji TAKAKI1 and Nobutaka UEDA2 1Kobe Design University, Nishi-ku, Kobe, Hyogo 651-2196, Japan 2Hiroshima-Gakuin, Nishi-ku, Hiroshima, Hiroshima 733-0875, Japan *E-mail address: [email protected] *E-mail address: [email protected] (Received November 3, 2006; Accepted August 10, 2007) Keywords: Spiral, Curvature, Logarithmic Spiral, Archimedean Spiral, Vortex Abstract. A method is proposed to characterize and classify shapes of plane spirals, and it is applied to some spiral patterns from historical monuments and vortices observed in an experiment. The method is based on a relation between the length variable along a curve and the radius of its local curvature. Examples treated in this paper seem to be classified into four types, i.e. those of the Archimedean spiral, the logarithmic spiral, the elliptic vortex and the hyperbolic spiral. 1. Introduction Spiral patterns are seen in many cultures in the world from ancient ages. They give us a strong impression and remind us of energy of the nature. Human beings have been familiar to natural phenomena and natural objects with spiral motions or spiral shapes, such as swirling water flows, swirling winds, winding stems of vines and winding snakes. It is easy to imagine that powerfulness of these phenomena and objects gave people a motivation to design spiral shapes in monuments, patterns of cloths and crafts after spiral shapes observed in their daily lives. Therefore, it would be reasonable to expect that spiral patterns in different cultures have the same geometrical properties, or at least are classified into a small number of common types. -

Superconics + Spirals 17 July 2017 1 Superconics + Spirals Cye H
Superconics + Spirals 17 July 2017 Superconics + Spirals Cye H. Waldman Copyright 2017 Introduction Many spirals are based on the simple circle, although modulated by a variable radius. We have generalized the concept by allowing the circle to be replaced by any closed curve that is topologically equivalent to it. In this note we focus on the superconics for the reasons that they are at once abundant, analytical, and aesthetically pleasing. We also demonstrate how it can be applied to random closed forms. The spirals of interest are of the general form . A partial list of these spirals is given in the table below: Spiral Remarks Logarithmic b = flair coefficient m = 1, Archimedes Archimedean m = 2, Fermat (generalized) m = -1, Hyperbolic m = -2, Lituus Parabolic Cochleoid Poinsot Genesis of the superspiral (1) The idea began when someone on line was seeking to create a 3D spiral with a racetrack planiform. Our first thought was that the circle readily transforms to an ellipse, for example Not quite a racetrack, but on the right track. The rest cascades in a hurry, as This is subject to the provisions that the random form is closed and has no crossing lines. This also brought to mind Euler’s famous derivation, connecting the five most famous symbols in mathematics. 1 Superconics + Spirals 17 July 2017 In the present paper we’re concerned primarily with the superconics because the mapping is well known and analytic. More information on superconics can be found in Waldman (2016) and submitted for publication by Waldman, Chyau, & Gray (2017). Thus, we take where is a parametric variable (not to be confused with the polar or tangential angles). -
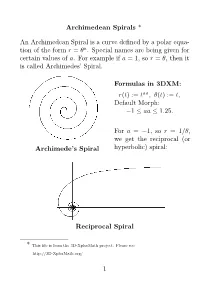
Archimedean Spirals * an Archimedean Spiral Is a Curve
Archimedean Spirals * An Archimedean Spiral is a curve defined by a polar equa- tion of the form r = θa. Special names are being given for certain values of a. For example if a = 1, so r = θ, then it is called Archimedes’ Spiral. Formulas in 3DXM: r(t) := taa, θ(t) := t, Default Morph: 1 aa 1.25. − ≤ ≤ For a = 1, so r = 1/θ, we get the− reciprocal (or Archimede’s Spiral hyperbolic) spiral: Reciprocal Spiral * This file is from the 3D-XplorMath project. Please see: http://3D-XplorMath.org/ 1 The case a = 1/2, so r = √θ, is called the Fermat (or hyperbolic) spiral. Fermat’s Spiral While a = 1/2, or r = 1/√θ, it is called the Lituus: − Lituus 2 In 3D-XplorMath, you can change the parameter a by go- ing to the menu Settings Set Parameters, and change the value of aa. You can see→an animation of Archimedean spirals where the exponent a = aa varies gradually, be- tween 1 and 1.25. See the Animate Menu, entry Morph. − The reason that the parabolic spiral and the hyperbolic spiral are so named is that their equations in polar coor- dinates, rθ = 1 and r2 = θ, respectively resembles the equations for a hyperbola (xy = 1) and parabola (x2 = y) in rectangular coordinates. The hyperbolic spiral is also called reciprocal spiral be- cause it is the inverse curve of Archimedes’ spiral, with inversion center at the origin. The inversion curve of any Archimedean spirals with re- spect to a circle as center is another Archimedean spiral, scaled by the square of the radius of the circle. -

APS March Meeting 2012 Boston, Massachusetts
APS March Meeting 2012 Boston, Massachusetts http://www.aps.org/meetings/march/index.cfm i Monday, February 27, 2012 8:00AM - 11:00AM — Session A51 DCMP DFD: Colloids I: Beyond Hard Spheres Boston Convention Center 154 8:00AM A51.00001 Photonic Droplets Containing Transparent Aqueous Colloidal Suspensions with Optimal Scattering Properties JIN-GYU PARK, SOFIA MAGKIRIADOU, Department of Physics, Harvard University, YOUNG- SEOK KIM, Korea Electronics Technology Institute, VINOTHAN MANOHARAN, Department of Physics, Harvard University, HARVARD UNIVERSITY TEAM, KOREA ELECTRONICS TECHNOLOGY INSTITUTE COLLABORATION — In recent years, there has been a growing interest in quasi-ordered structures that generate non-iridescent colors. Such structures have only short-range order and are isotropic, making colors invariant with viewing angle under natural lighting conditions. Our recent simulation suggests that colloidal particles with independently controlled diameter and scattering cross section can realize the structural colors with angular independence. In this presentation, we are exploiting depletion-induced assembly of colloidal particles to create isotropic structures in a milimeter-scale droplet. As a model colloidal particle, we have designed and synthesized core-shell particles with a large, low refractive index shell and a small, high refractive index core. The remarkable feature of these particles is that the total cross section for the entire core-shell particle is nearly the same as that of the core particle alone. By varying the characteristic length scales of the sub-units of such ‘photonic’ droplet we aim to tune wavelength selectivity and enhance color contrast and viewing angle. 8:12AM A51.00002 Curvature-Induced Capillary Interaction between Spherical Particles at a Liquid Interface1 , NESRIN SENBIL, CHUAN ZENG, BENNY DAVIDOVITCH, ANTHONY D. -

John Sharp Spirals and the Golden Section
John Sharp Spirals and the Golden Section The author examines different types of spirals and their relationships to the Golden Section in order to provide the necessary background so that logic rather than intuition can be followed, correct value judgments be made, and new ideas can be developed. Introduction The Golden Section is a fascinating topic that continually generates new ideas. It also has a status that leads many people to assume its presence when it has no relation to a problem. It often forces a blindness to other alternatives when intuition is followed rather than logic. Mathematical inexperience may also be a cause of some of these problems. In the following, my aim is to fill in some gaps, so that correct value judgements may be made and to show how new ideas can be developed on the rich subject area of spirals and the Golden Section. Since this special issue of the NNJ is concerned with the Golden Section, I am not describing its properties unless appropriate. I shall use the symbol I to denote the Golden Section (I |1.61803 ). There are many aspects to Golden Section spirals, and much more could be written. The parts of this paper are meant to be read sequentially, and it is especially important to understand the different types of spirals in order that the following parts are seen in context. Part 1. Types of Spirals In order to understand different types of Golden Section spirals, it is necessary to be aware of the properties of different types of spirals. This section looks at spirals from that viewpoint. -

Curves Defined Using Polar Coordinates
Curves defined using polar coordinates Instead of using Cartesian (x; y) coordinates, we can describe points in the plane by specifying how far they are from some fixed reference point (the pole) and the angle the line from the point to the pole makes with some reference direction. The convention is to treat the origin (0; 0) as the pole, and the reference direction as the positive x-axis. In this way, we can convert back and forth between polar coordinates and cartesian. So a point in the plane may be specified by a pair (r; θ) like this: (r; θ) r θ Here are some points in both xy-coordinates and polar coordinates: Cartesian polar (1; 0) (p1; 0) (1; 1) ( 2; π p 4 (0; 1) (1; π2 (−1; 0) (p1; π) 5π (−1; −1 ( 2; 4 3π (0; −1) (1; 2 (3; 7) (7:6157:::; 1:1659:::) (0:5673:::; −1:9178) (2; 5) To convert back and forth, we can use these relationships: x = r cos θ; y = r sin θ and p y r = x 2 + y 2; θ = tan−1 x with the caveat that the theta value yielded might be negative. For example, (r; θ); (−r; θ + π); (−r; θ + 3π); (r; θ + 2π) all represent the same point. This makes thing interesting: simple polar equations can create complex curves. Which brings us the main oddity of polar coordinates: they are not unique. If we add 2π (or any multiple of 2 pi) to θ, we are specifying the same point. We can even use a negative r if we add an odd multiple π to θ. -

Arm Spiral Antennas
INVESTIGATION OF CYLINDRICALLY-CONFORMED FOUR- ARM SPIRAL ANTENNAS A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Engineering By DOUGLAS JEREMY GLASS B.S.E.P., Wright State University, 2003 2007 Wright State University WRIGHT STATE UNIVERSITY SCHOOL OF GRADUATE STUDIES June 29, 2007 I HEREBY RECOMMEND THAT THE THESIS PREPARED UNDER MY SUPERVISION BY Douglas Jeremy Glass ENTITLED Investigation of Cylindrically- Conformed Four-Arm Spiral Antennas BE ACCEPTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science in Engineering. _________________________________ Ronald Riechers, Ph.D. Thesis Director _________________________________ Fred Garber, Ph.D. Department Chair Committee on Final Examination _________________________________ Ronald Riechers, Ph.D. _________________________________ Fred Garber, Ph.D. _________________________________ Marian Kazimierczuk, Ph.D. _________________________________ Joseph F. Thomas, Jr., Ph.D. Dean, School of Graduate Studies ABSTRACT Glass, Douglas Jeremy, M.S. Egr., Department of Electrical Engineering, Wright State University, 2007. Investigation of Cylindrically-Conformed Four-Arm Spiral Antennas. A four-arm spiral antenna offers broadband frequency response, wide beamwidths, reduced size compared to other antenna designs, and the ability to determine the relative direction of an incident signal with appropriate mode-forming. The reduced overall area projection of the four-arm spiral antenna compared to other antenna designs -

The Mathematics and Art of Spirals Workshop
Proceedings of Bridges 2013: Mathematics, Music, Art, Architecture, Culture The Mathematics and Art of Spirals Workshop Ann Hanson Science and Mathematics Department, Columbia College Chicago, 600 S. Michigan, Chicago, IL 606005 [email protected] Abstract In this workshop, the participants will learn several properties of spirals. Some of the properties are: that all spirals start from a fixed point and move out from that point in a regular manner. Other universal properties of spirals and their mathematical connections will be demonstrated by making an Archimedean spiral, a Golden spiral and a Baravelle spiral. The presenter will supply all equipment needed for the activities. A spiral is a curve that moves out from the center in a regular manner getting progressively farther away as it moves out. The spiral shape is so prevalent that it is easy not to notice them. They can be seen in natural objects, such as in the shells of mollusks, sunflower heads and in vortices in air and water. Spirals are also seen in the coil of watch balance springs, the grooves of a phonograph record and in the roll of paper towels. In addition, all spirals have some common characteristics. They all have a fixed starting point (center) and move out from the center in a regular manner. Spirals grow by self-accumulation and solve a problem of efficient growth in terms of energy, materials and expansion within space. The previously mentioned properties as well as other mathematical properties will be examined and demonstrated through hands-on activities. For example, the participants will make a one type of spiral called an Archimedean spiral using polar graph paper. -

Collected Atos
Mathematical Documentation of the objects realized in the visualization program 3D-XplorMath Select the Table Of Contents (TOC) of the desired kind of objects: Table Of Contents of Planar Curves Table Of Contents of Space Curves Surface Organisation Go To Platonics Table of Contents of Conformal Maps Table Of Contents of Fractals ODEs Table Of Contents of Lattice Models Table Of Contents of Soliton Traveling Waves Shepard Tones Homepage of 3D-XPlorMath (3DXM): http://3d-xplormath.org/ Tutorial movies for using 3DXM: http://3d-xplormath.org/Movies/index.html Version November 29, 2020 The Surfaces Are Organized According To their Construction Surfaces may appear under several headings: The Catenoid is an explicitly parametrized, minimal sur- face of revolution. Go To Page 1 Curvature Properties of Surfaces Surfaces of Revolution The Unduloid, a Surface of Constant Mean Curvature Sphere, with Stereographic and Archimedes' Projections TOC of Explicitly Parametrized and Implicit Surfaces Menu of Nonorientable Surfaces in previous collection Menu of Implicit Surfaces in previous collection TOC of Spherical Surfaces (K = 1) TOC of Pseudospherical Surfaces (K = −1) TOC of Minimal Surfaces (H = 0) Ward Solitons Anand-Ward Solitons Voxel Clouds of Electron Densities of Hydrogen Go To Page 1 Planar Curves Go To Page 1 (Click the Names) Circle Ellipse Parabola Hyperbola Conic Sections Kepler Orbits, explaining 1=r-Potential Nephroid of Freeth Sine Curve Pendulum ODE Function Lissajous Plane Curve Catenary Convex Curves from Support Function Tractrix