Redalyc.An Approach to the Biological, Historical and Psychological
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Disorders in Premature Ovarian Failure
Human Reproduction Update, Vol.8, No.4 pp. 483±491, 2002 Genetic disorders in premature ovarian failure T.Laml1,3, O.Preyer1, W.Umek1, M.HengstschlaÈger2 and E.Hanzal1 University of Vienna Medical School, Department of Obstetrics and Gynaecology, 1Division of Gynaecology and 2Division of Prenatal Diagnosis and Therapy, Waehringer Guertel 18-20, A-1090 Vienna, Austria 3To whom correspondence should be addressed. E-mail: [email protected] This review presents the genetic disorders associated with premature ovarian failure (POF), obtained by Medline, the Cochrane Library and hand searches of pertinent references of English literature on POF and genetic determinants cited between the year 1966 and February 2002. X monosomy or X deletions and translocations are known to be responsible for POF. Turner's syndrome, as a phenotype associated with complete or partial monosomy X, is linked to ovarian failure. Among heterozygous carriers of the fragile X mutation, POF was noted as an unexpected phenotype in the early 1990s. Autosomal disorders such as mutations of the phosphomannomutase 2 (PMM2) gene, the galactose-1-phosphate uridyltransferase (GALT) gene, the FSH receptor (FSHR) gene, chromosome 3q containing the Blepharophimosis gene and the autoimmune regulator (AIRE) gene, responsible for polyendocrinopathy-candidiasis-ectodermal dystrophy, have been identi®ed in patients with POF. In conclusion, the relationship between genetic disorders and POF is clearly demonstrated in this review. Therefore, in the case of families affected by POF a thorough screening, including cytogenetic analysis, should be performed. Key words: autosomal disorders/FSH receptor/inhibin/premature ovarian failure/X chromosome abnormalities TABLE OF CONTENTS diagnosis requires histological examination of a full-thickness ovarian biopsy (Metha et al., 1992; Olivar, 1996). -

Turner Syndrome Diagnosed in Northeastern Malaysia Kannan T P, Azman B Z, Ahmad Tarmizi a B, Suhaida M A, Siti Mariam I, Ravindran A, Zilfalil B A
Original Article Singapore Med J 2008; 49(5) : 400 Turner syndrome diagnosed in northeastern Malaysia Kannan T P, Azman B Z, Ahmad Tarmizi A B, Suhaida M A, Siti Mariam I, Ravindran A, Zilfalil B A ABSTRACT counselling, gonadal dysgenesis, short stature, Introduction: Turner syndrome affects about one Turner syndrome in 2,000 live-born females, and the wide range Singapore Med J 2008; 49(5): 400-404 of somatic features indicates that a number of different X-located genes are responsible for the INTRODUCTION complete phenotype. This retrospective study Turner syndrome, gonadal dysgenesis or gonadal agenesis highlights the Turner syndrome cases confirmed represents a special variant of hypergonadotrophic through cytogenetic analysis at the Human hypogonadism, and is due to the lack of the second sex Genome Centre of Universiti Sains Malaysia, from chromosome or parts of it. This syndrome affects about one 2001 to 2006. in 2,000 live-born females.(1) The wide range of somatic features in Turner syndrome indicates that a number of Methods: Lymphocyte cultures were set up using different X-located genes are responsible for the complete peripheral blood samples, chromosomes were phenotype.(2) The syndrome includes those individuals prepared, G-banded, karyotyped and analysed in with a phenotypic spectrum from female to male, with accordance to guidelines from the International varying clinical stigmata of the syndrome, as described System for Human Cytogenetic Nomenclature. by Turner.(3) Though many karyotype abnormalities have been described in association with Turner syndrome, Results: The various karyotype patterns observed monoclonal monosomy X and its various mosaicisms, Human Genome were 45,X; 46,X,i,(Xq); 45,X/45,X,+mar; each with an X monosomic (XO) cell clone, are the most Centre, Universiti Sains 45,X/46,X,i,(Xq) and 45,X/46,XY. -

Orphanet Report Series Rare Diseases Collection
Marche des Maladies Rares – Alliance Maladies Rares Orphanet Report Series Rare Diseases collection DecemberOctober 2013 2009 List of rare diseases and synonyms Listed in alphabetical order www.orpha.net 20102206 Rare diseases listed in alphabetical order ORPHA ORPHA ORPHA Disease name Disease name Disease name Number Number Number 289157 1-alpha-hydroxylase deficiency 309127 3-hydroxyacyl-CoA dehydrogenase 228384 5q14.3 microdeletion syndrome deficiency 293948 1p21.3 microdeletion syndrome 314655 5q31.3 microdeletion syndrome 939 3-hydroxyisobutyric aciduria 1606 1p36 deletion syndrome 228415 5q35 microduplication syndrome 2616 3M syndrome 250989 1q21.1 microdeletion syndrome 96125 6p subtelomeric deletion syndrome 2616 3-M syndrome 250994 1q21.1 microduplication syndrome 251046 6p22 microdeletion syndrome 293843 3MC syndrome 250999 1q41q42 microdeletion syndrome 96125 6p25 microdeletion syndrome 6 3-methylcrotonylglycinuria 250999 1q41-q42 microdeletion syndrome 99135 6-phosphogluconate dehydrogenase 67046 3-methylglutaconic aciduria type 1 deficiency 238769 1q44 microdeletion syndrome 111 3-methylglutaconic aciduria type 2 13 6-pyruvoyl-tetrahydropterin synthase 976 2,8 dihydroxyadenine urolithiasis deficiency 67047 3-methylglutaconic aciduria type 3 869 2A syndrome 75857 6q terminal deletion 67048 3-methylglutaconic aciduria type 4 79154 2-aminoadipic 2-oxoadipic aciduria 171829 6q16 deletion syndrome 66634 3-methylglutaconic aciduria type 5 19 2-hydroxyglutaric acidemia 251056 6q25 microdeletion syndrome 352328 3-methylglutaconic -

Intersex 101
INTERSEX 101 With Your Guide: Phoebe Hart Secretary, AISSGA (Androgen Insensitivity Syndrome Support Group, Australia) And all‐round awesome person! WHAT IS INTERSEX? • a range of biological traits or variations that lie between “male” and “female”. • chromosomes, genitals, and/or reproductive organs that are traditionally considered to be both “male” and “female,” neither, or atypical. • 1.7 – 2% occurrence in human births REFERENCE: Australians Born with Atypical Sex Characteristics: Statistics & stories from the first national Australian study of people with intersex variations 2015 (in press) ‐ Tiffany Jones, School of Education, University of New England (UNE), Morgan Carpenter, OII Australia, Bonnie Hart, Androgyn Insensitivity Syndrome Support Group Australia (AISSGA) & Gavi Ansara, National LGBTI Health Network XY CHROMOSOMES ..... Complete Androgen Insensitivity Syndrome (CAIS) ..... Partial Androgen Insensitivity Syndrome (PAIS) ..... 5‐alpha‐reductase Deficiency (5‐ARD) ..... Swyer Syndrome/ Mixed Gonadal Dysgenesis (MGD) ..... Leydig Cell Hypoplasia ..... Persistent Müllerian Duct Syndrome ..... Hypospadias, Epispadias, Aposthia, Micropenis, Buried Penis, Diphallia ..... Polyorchidism, Cryptorchidism XX CHROMOSOMES ..... de la Chapelle/XX Male Syndrome ..... MRKH/Vaginal (or Müllerian) agenesis ..... XX Gonadal Dysgenesis ..... Uterus Didelphys ..... Progestin Induced Virilization XX or XY CHROMOSOMES ...... Congenital Adrenal Hyperplasia (CAH) ..... Ovo‐testes (formerly called "true hermaphroditism") .... -

From Gene to Gender - What We’Ve Learned and What We Need to Learn - New Prospects in DSD Research
3rd International Symposium on Disorders of Sex Development 20 – 22 May 2011 · Universität zu Lübeck Programme & Abstracts From Gene to Gender - What we’ve learned and what we need to learn - New Prospects in DSD Research Sessions on Genetics, Hormones & Action, Morphologic Variation in DSD and Clinical Aspects · Round Table · Poster Session and Oral Sessions 20 – 22 May 2011, Lübeck, Germany 1 Contents Contents...................................................................................................................................1 Welcome Remarks ...................................................................................................................3 About EuroDSD........................................................................................................................4 General Information..................................................................................................................5 Scientific Programme ...............................................................................................................6 Social Programme....................................................................................................................9 Speakers’ List.........................................................................................................................10 Poster Presentation................................................................................................................12 Abstracts Keynote Lectures ...................................................................................................15 -

Endocrine Test Selection and Interpretation
The Quest Diagnostics Manual Endocrinology Test Selection and Interpretation Fourth Edition The Quest Diagnostics Manual Endocrinology Test Selection and Interpretation Fourth Edition Edited by: Delbert A. Fisher, MD Senior Science Officer Quest Diagnostics Nichols Institute Professor Emeritus, Pediatrics and Medicine UCLA School of Medicine Consulting Editors: Wael Salameh, MD, FACP Medical Director, Endocrinology/Metabolism Quest Diagnostics Nichols Institute San Juan Capistrano, CA Associate Clinical Professor of Medicine, David Geffen School of Medicine at UCLA Richard W. Furlanetto, MD, PhD Medical Director, Endocrinology/Metabolism Quest Diagnostics Nichols Institute Chantilly, VA ©2007 Quest Diagnostics Incorporated. All rights reserved. Fourth Edition Printed in the United States of America Quest, Quest Diagnostics, the associated logo, Nichols Institute, and all associated Quest Diagnostics marks are the trademarks of Quest Diagnostics. All third party marks − ®' and ™' − are the property of their respective owners. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopy, recording, and information storage and retrieval system, without permission in writing from the publisher. Address inquiries to the Medical Information Department, Quest Diagnostics Nichols Institute, 33608 Ortega Highway, San Juan Capistrano, CA 92690-6130. Previous editions copyrighted in 1996, 1998, and 2004. Re-order # IG1984 Forward Quest Diagnostics Nichols Institute has been -

'Control of Sex Development'
Biason-Lauber, A (2010). Control of sex development. Best Practice and Research Clinical Endocrinology & Metabolism, 24(2):163-86. Postprint available at: http://www.zora.uzh.ch University of Zurich Posted at the Zurich Open Repository and Archive, University of Zurich. Zurich Open Repository and Archive http://www.zora.uzh.ch Originally published at: Best Practice and Research Clinical Endocrinology & Metabolism 2010, 24(2):163-86. Winterthurerstr. 190 CH-8057 Zurich http://www.zora.uzh.ch Year: 2010 Control of sex development Biason-Lauber, A Biason-Lauber, A (2010). Control of sex development. Best Practice and Research Clinical Endocrinology & Metabolism, 24(2):163-86. Postprint available at: http://www.zora.uzh.ch Posted at the Zurich Open Repository and Archive, University of Zurich. http://www.zora.uzh.ch Originally published at: Best Practice and Research Clinical Endocrinology & Metabolism 2010, 24(2):163-86. CONTROL OF SEX DEVELOPMENT Anna Biason-Lauber University Children’s Hospital, Zurich, Switzerland Correspondence to : Anna Biason-Lauber, MD University Children’s Hospital Div. of Endocrinology/Diabetology Steinwiesstrasse 75 CH-8032 Zurich Switzerland Tel + 41 44 266 7623 Fax + 41 44 266 7169 E-mail: [email protected] 1 ABSTRACT The process of sexual differentiation is central for reproduction of almost all metazoan, and therefore for maintenance of practically all multicellular organisms. In sex development we can distinguish two different processes¸ sex determination , that is the developmental decision that directs the undifferentiated embryo into a sexually dimorphic individual. In mammals, sex determination equals gonadal development. The second process known as sex differentiation takes place once the sex determination decision has been made through factors produced by the gonads that determine the development of the phenotypic sex. -
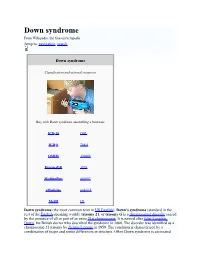
Down Syndrome from Wikipedia, the Free Encyclopedia Jump To: Navigation, Search
Down syndrome From Wikipedia, the free encyclopedia Jump to: navigation, search Down syndrome Classification and external resources Boy with Down syndrome assembling a bookcase ICD-10 Q 90. ICD-9 758.0 OMIM 190685 DiseasesDB 3898 MedlinePlus 000997 eMedicine ped/615 MeSH [2] Down syndrome (the most common term in US English), Down's syndrome (standard in the rest of the English-speaking world), trisomy 21, or trisomy G is a chromosomal disorder caused by the presence of all or part of an extra 21st chromosome. It is named after John Langdon Down, the British doctor who described the syndrome in 1866. The disorder was identified as a chromosome 21 trisomy by Jérôme Lejeune in 1959. The condition is characterized by a combination of major and minor differences in structure. Often Down syndrome is associated with some impairment of cognitive ability and physical growth as well as facial appearance. Down syndrome in a baby can be identified with amniocentesis during pregnancy or at birth. Individuals with Down syndrome tend to have a lower than average cognitive ability, often ranging from mild to moderate developmental disabilities. A small number have severe to profound mental disability. The incidence of Down syndrome is estimated at 1 per 800 to 1,000 births, although these statistics are heavily influenced by older mothers. Other factors may also play a role. Many of the common physical features of Down syndrome may also appear in people with a standard set of chromosomes, including microgenia (an abnormally small chin)[1], an unusually round face, macroglossia [2] (protruding or oversized tongue), an almond shape to the eyes caused by an epicanthic fold of the eyelid, upslanting palpebral fissures (the separation between the upper and lower eyelids), shorter limbs, a single transverse palmar crease (a single instead of a double crease across one or both palms, also called the Simian crease), poor muscle tone, and a larger than normal space between the big and second toes. -

Adolescent Girls with Pure Gonadal Dysgenesis: a Rare Disease J Sahaa, K Begumb, KA Khanomc, I Prasadd, S Aktere
Journal of Bangladesh College of Physicians and Surgeons Vol. 36, No. 4, October 2018 Adolescent Girls with Pure Gonadal Dysgenesis: A Rare Disease J SAHAa, K BEGUMb, KA KHANOMc, I PRASADd, S AKTERe Summary: age of 16yr because of absence of secondary sexual Gonadal dysgenesis is a rare cause of primary characteristics as well as menarche of her daughter. amenorrhoea ,which is a relatively common problem among In both cases, blood test showed very high levels of follicle teenage girls.Primary amenorrhoea occurs in patient with stimulating hormone (FSH) & luteinizing hormone (LH), gonadal dysgenesis because of absence or limited ovarian low levels of oestradiol& very low level of AMH. USG function due to inappropriate development.Streak gonads findings of both cases showed a bit hypoplastic uterus and are unable to produce estrogens and/or androgens,resulting volume of ovaries were smaller than normal. A diagnostic in minimal to no development of secondary sexual laparoscopy with biopsy of both gonads of one case was characteristics.Adrenal androgens may induce production performed.Another case did not give consent for of pubic hair,but patient will have minimal breast laparoscopy.Hormonal replacement therapy was applied development.These patients may have a family history of on them for establishment of normal menstruation and infertility, short stature,sensorineural deafness,ataxia,mild mental retardation or gonadoblastoma. menstruation was established in both cases. Here two cases of primary amenorrhoea due to pure An early diagnosis is extremely important to prevent long gonadal dysgenesis are presented. 1st one was a 18yr old term consequences of Gonadal dysgenesis. girl whose mother consulted with a gynaecologist at the Key word: Primary Amenorrhoea, Gonadal dysgenesis. -

Endocrinology
THE AMERICAN BOARD OF PEDIATRICS® CONTENT OUTLINE Pediatric Endocrinology Subspecialty In-Training, Certification, and Maintenance of Certification (MOC) Examinations INTRODUCTION This document was prepared by the American Board of Pediatrics Subboard of Pediatric Endocrinology for the purpose of developing in-training, certification, and maintenance of certification examinations. The outline defines the body of knowledge from which the Subboard samples to prepare its examinations. The content specification statements located under each category of the outline are used by item writers to develop questions for the examinations; they broadly address the specific elements of knowledge within each section of the outline. Pediatric Endocrinology Each Pediatric Endocrinology exam is built to the same specifications, also known as the blueprint. This blueprint is used to ensure that, for the initial certification and in-training exams, each exam measures the same depth and breadth of content knowledge. Similarly, the blueprint ensures that the same is true for each Maintenance of Certification exam form. The table below shows the percentage of questions from each of the content domains that will appear on an exam. Please note that the percentages are approximate; actual content may vary. Initial Maintenance Certificatio of Content Categories n and Certification In-Training (MOC) 1. Carbohydrate Metabolism 16% 16% 2. Bone and Mineral Metabolism 8% 8% Thyroid Hormones (Thyroxine 3. [T4] and Triiodothyronine [T3]) 13% 14% 4. Adrenal Disorders 12% 12% 5. Pituitary/Hypothalamus 10% 10% 6. Growth 12% 14% 7. Reproductive Endocrine System 12% 12% 8. Other Hormones 3% 3% 9. Lipoproteins and Lipids 3% 3% Multiple endocrine neoplasia and 10. polyglandular autoimmune disease 2% 2% 11. -

Genetic Aspects of Premature Ovarian Failure Wieacker P J
Journal für Reproduktionsmedizin und Endokrinologie – Journal of Reproductive Medicine and Endocrinology – Andrologie • Embryologie & Biologie • Endokrinologie • Ethik & Recht • Genetik Gynäkologie • Kontrazeption • Psychosomatik • Reproduktionsmedizin • Urologie Genetic Aspects of Premature Ovarian Failure Wieacker P J. Reproduktionsmed. Endokrinol 2009; 6 (1), 17-18 www.kup.at/repromedizin Online-Datenbank mit Autoren- und Stichwortsuche Offizielles Organ: AGRBM, BRZ, DVR, DGA, DGGEF, DGRM, D·I·R, EFA, OEGRM, SRBM/DGE Indexed in EMBASE/Excerpta Medica/Scopus Krause & Pachernegg GmbH, Verlag für Medizin und Wirtschaft, A-3003 Gablitz FERRING-Symposium digitaler DVR 2021 Mission possible – personalisierte Medizin in der Reproduktionsmedizin Was kann die personalisierte Kinderwunschbehandlung in der Praxis leisten? Freuen Sie sich auf eine spannende Diskussion auf Basis aktueller Studiendaten. SAVE THE DATE 02.10.2021 Programm 12.30 – 13.20Uhr Chair: Prof. Dr. med. univ. Georg Griesinger, M.Sc. 12:30 Begrüßung Prof. Dr. med. univ. Georg Griesinger, M.Sc. & Dr. Thomas Leiers 12:35 Sind Sie bereit für die nächste Generation rFSH? Im Gespräch Prof. Dr. med. univ. Georg Griesinger, Dr. med. David S. Sauer, Dr. med. Annette Bachmann 13:05 Die smarte Erfolgsformel: Value Based Healthcare Bianca Koens 13:15 Verleihung Frederik Paulsen Preis 2021 Wir freuen uns auf Sie! Premature Ovarian Failure Genetic Aspects of Premature Ovarian Failure P. Wieacker Premature ovarian failure (POF) is characterized by the combination of amenorrhea before age 40 years and hypergonadotropic hypogonadism. The prevalence is approximately 1 %. Genetic causes of POF include chromosome aberrations and monogenic defects. Furthermore, polygenic-multifactorial inheritance can be assumed in a subset of cases. J Reproduktionsmed Endokrinol 2009; 6 (1): 17–8. -

Gene Analysis in Patients with Premature Ovarian Failure Or
Maturitas 57 (2007) 399–404 Gene analysis in patients with premature ovarian failure or gonadal dysgenesis: A preliminary study Ana Maria Massad-Costa a, Ismael Dale Cotrim Guerreiro da Silva a, Regina Affonso a, Jose´ Maria Soares Jr. a,∗,Marcia´ Gaspar Nunes a, Geraldo Rodrigues de Lima a, Edmund C. Baracat a,b a Department of Gynecology, Federal University of S˜ao Paulo, Escola Paulista de Medicina, Brazil b Department of Gynecology and Obstetrics, University of S˜ao Paulo, Brazil Received 12 November 2006; received in revised form 18 March 2007; accepted 22 April 2007 Abstract Objective: The aim of this study was to evaluate the presence of mutations in the coding region of the QM gene and fragile X in patients with premature ovarian failure and gonadal dysgenesis. Methods: After approval by the local Ethics Committee, blood samples, in EDTA, of 100 normally ovulating women, 23 with premature ovarian failure (POF) and 14 with gonadal dysgenesis 46XX, aged less than 40 years, were screened for mutation in the QM gene coding region. All patients with POF have 46, XX karyotype and serum levels of follicle-stimulating hormone (FSH) over 30 mIU/mL. In addition, all samples from patients with premature ovarian failure underwent analysis for fragile X. Results: The QM gene located at a hotspot region (Xq28) showed five points of mutations in a patient with premature ovarian failure. Four of them were able to change the amino acid sequence of the protein. None of our patients were diagnosed as having pre or mutant X fragile syndrome. Conclusion: Our study suggests that Xq28 (QM gene) may be involved in ovary failure.