Role of Lymphotoxin Beta and Cell Adhesion Molecule (CEACAM1) in Innate and Adaptive Immune Activation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Combination Immune Therapy
Combination Immune Therapy Translational Medicine Plenary SWOG April 17, 2017 San Francisco T‐cell Activation, Proliferation, and Function is Controlled by Multiple Agonist and Antagonist Signals 1. Co‐stimulation via CD28 ligation 2. CTLA‐4 ligation on activated T 3. T cell function in tissue is subject transduces T cell activating signals cells down‐regulates T cell to feedback inhibition responses T cell Functional Block T cell Activation T cell Activation T cell Proliferative Block T cell T cell APC T cell CTLA‐4 T cell CTLA‐4 TCR TCR PD‐1 PD‐1 PD‐1 IFN‐gamma TCR CD28 Cytokines TCR CD4 MHC MHC PD‐‐L1 MHC B7 MHC CD28 B7 PD‐‐L1 Tumor or Tumor or APC APC Immune cell B7 immune cell APC NK Presence of PD-L1 or TILs1 PPD-L1 /TIL+ PD-L1/TIL + + PD-L1 /TIL+ + PD-L1 /TIL PD-L1 /TIL D-L1 /TIL+ Schalper and Rimm, Yale University NSCLC PD-L1/TIL 45% 17% 26% 12% Type 1 Type 2 Type 3 Type 4 45% 41% 13% 1% Melanoma45% 17% 26% 12% Taube et al Type 1 Type 2 Type 3 Type 4 Tumor‐specific T cells are contained in the PD‐1+ TIL population and are functional after in vitro culture Spectrum of PD‐1/PD‐L1 Antagonist Activity Active • Melanoma Minimal to no activity • Renal cancer (clear cell and non‐clear cell) • Prostate cancer • NSCLC – adenocarcinoma and squamous cell • MMR+ (MSS) colon cancer • Small cell lung cancer • Myeloma • Head and neck cancer • Pancreatic cancer • Gastric and gastroesophageal junction • MMR‐repair deficient tumors (colon, cholangiocarcinoma) • Bladder • Triple negative breast cancer • Ovarian Major PD‐1/PD‐L1 antagonists • Hepatocellular -
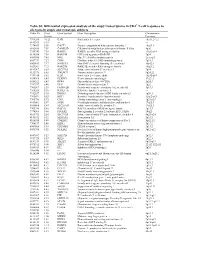
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3 -

Bioinformatics Method Identifies Potential Biomarkers of Dilated Cardiomyopathy in a Human Induced Pluripotent Stem Cell‑Derived Cardiomyocyte Model
EXPERIMENTAL AND THERAPEUTIC MEDICINE 14: 2771-2778, 2017 Bioinformatics method identifies potential biomarkers of dilated cardiomyopathy in a human induced pluripotent stem cell‑derived cardiomyocyte model YU ZHUANG1, YU-JIA GONG2, BEI-FEN ZHONG3, YI ZHOU1 and LI GONG4 1Department of Cardiovascular Surgery, Shanghai First People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080; 2Stomatology Faculty, School of Medicine, Nantong University, Nantong, Jiangsu 226000; 3Department of Urology, Shanghai First People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080; 4Department of Cardiothoracic Surgery, The Affiliated Huai'an Hospital of Xuzhou Medical University, Huai'an, Jiangsu 223002, P.R. China Received February 19, 2016; Accepted February 10, 2017 DOI: 10.3892/etm.2017.4850 Abstract. Dilated cardiomyopathy (DCM) is the most common crucial nodes in module 2, which were linked to each other. type of cardiomyopathy that account for the majority of heart In conclusion, several potential biomarkers for DCM were failure cases. The present study aimed to reveal the under- identified, such as MMP2, FLT1, CDH1, ITGB6, COL6A3, lying molecular mechanisms of DCM and provide potential COL6A1, LAMC2, PENK and APLNR. These genes may serve biomarkers for detection of this condition. The public dataset significant roles in DCM via involvement of various BPs, such of GSE35108 was downloaded, and 4 normal induced pluripo- as blood vessel and vasculature development and cell adhe- tent stem cell (iPSC)-derived cardiomyocytes (N samples) and sion, and the ECM-receptor interaction pathway. 4 DCM iPSC-derived cardiomyocytes (DCM samples) were utilized. Raw data were preprocessed, followed by identifica- Introduction tion of differentially expressed genes (DEGs) between N and DCM samples. -

(Lilrs) on Human Neutrophils: Modulators of Infection and Immunity
MINI REVIEW published: 13 May 2020 doi: 10.3389/fimmu.2020.00857 Leukocyte Immunoglobulin-Like Receptors (LILRs) on Human Neutrophils: Modulators of Infection and Immunity Alexander L. Lewis Marffy and Alex J. McCarthy* MRC Centre for Molecular Bacteriology and Infection, Imperial College London, London, United Kingdom Neutrophils have a crucial role in defense against microbes. Immune receptors allow neutrophils to sense their environment, with many receptors functioning to recognize signs of infection and to promote antimicrobial effector functions. However, the neutrophil Edited by: Nicole Thielens, response must be tightly regulated to prevent excessive inflammation and tissue damage, UMR5075 Institut de Biologie and regulation is achieved by expression of inhibitory receptors that can raise activation Structurale (IBS), France thresholds. The leukocyte immunoglobulin-like receptor (LILR) family contain activating Reviewed by: and inhibitory members that can up- or down-regulate immune cell activity. New ligands Debby Burshtyn, University of Alberta, Canada and functions for LILR continue to emerge. Understanding the role of LILR in neutrophil Tamás Laskay, biology is of general interest as they can activate and suppress antimicrobial responses University of Lübeck, Germany of neutrophils and because several human pathogens exploit these receptors for immune *Correspondence: Alex J. McCarthy evasion. This review focuses on the role of LILR in neutrophil biology. We focus on the [email protected] current knowledge of LILR expression -

Supplemental Text and Figures
Supplemental Materials and Methods Experimental bone metastasis assay Primary PCa cells were sorted by GFP marker from mTmG+ tumors, and 105 cells in 20μL PBS were injected using Hamilton syringe into the tibia of 6-week old NSG mice. Mice were monitored biweekly for moribund signs for euthanasia and organ harvest. Noninvasive mouse and ex vivo imaging MRI imaging with Bruker ICON and fluorescence imaging of fresh organs with metastasis enumeration were recently described (Lu et al. 2017). Primary prostate cell sphere formation assay Isolate of primary cells from prostate, culture and counting of prostatospheres on Matrigel were performed as described (Lukacs et al. 2010). For organoid culture assay, we followed a published matrigel embedding method (Chua et al. 2014). RNA-Seq and differential gene expression Total RNA was isolated from prostate tumors using Direct-zol RNA MiniPrep Kit (Zymo Research) and processed for stranded total RNA-Seq using Illumina HiSeq 4000 at Sequencing and Microarray Facility at MD Anderson Cancer Center. The differential expression analysis was performed using the DESeq2 package of R. P-values obtained after multiple binomial tests were adjusted using BH method. Significant genes are defined by using a cut-off of 0.05 on the BH corrected p-value and an absolute log2 fold change value of at least 1.5. Histology and western blot H&E stain, immunohistochemical (IHC) and western blot were performed as previously described (Ding et al. 2011; Wang et al. 2016). Primary antibodies for IHC include Ki67 (Fisher, RM-9106-S1), cleaved caspase 3 (Cell Signaling Technology aka CST, 9661), cyclin D1 (Fisher, clone SP4), TGFBR2 (Abcam, ab61213), BMPR2 (Abcam, ab130206), AR (EMD Millipore, 06-680), phospho- Akt (CST, 4060), GFP (CST, 2956), E-Cadherin (CST, 14472). -

CEACAM1 As a Multi-Purpose Target for Cancer Immunotherapy
CEACAM1 as a multi-purpose target for cancer immunotherapy The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Dankner, Matthew, Scott D. Gray-Owen, Yu-Hwa Huang, Richard S. Blumberg, and Nicole Beauchemin. 2017. “CEACAM1 as a multi- purpose target for cancer immunotherapy.” Oncoimmunology 6 (7): e1328336. doi:10.1080/2162402X.2017.1328336. http:// dx.doi.org/10.1080/2162402X.2017.1328336. Published Version doi:10.1080/2162402X.2017.1328336 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:34375168 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA ONCOIMMUNOLOGY 2017, VOL. 6, NO. 7, e1328336 (16 pages) https://doi.org/10.1080/2162402X.2017.1328336 REVIEW CEACAM1 as a multi-purpose target for cancer immunotherapy Matthew Dankner a, Scott D. Gray-Owen b, Yu-Hwa Huangc, Richard S. Blumberg c, and Nicole Beauchemin a aGoodman Cancer Research Centre, McGill University, Montreal, QC, Canada; bDepartment of Molecular Genetics, University of Toronto, Toronto, ON, Canada; cDivision of Gastroenterology, Hepatology, and Endoscopy, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA ABSTRACT ARTICLE HISTORY CEACAM1 is an extensively studied cell surface molecule with established functions in multiple cancer Received 17 February 2017 types, as well as in various compartments of the immune system. Due to its multi-faceted role as a Revised 3 May 2017 recently appreciated immune checkpoint inhibitor and tumor marker, CEACAM1 is an attractive target for Accepted 5 May 2017 cancer immunotherapy. -

Gastrointestinal Poorly-Differentiated and Anaplastic Carcinomas
ANNUAL MEETING ABSTRACTS 95A due to chronic androgen deficiency or to the genetic abnormality per se remains to be neoplastic follicular cells that demonstrated immunoreactivity for SDF-1 was scored as elucidated. Molecular genetic studies are required to obtain deeper insight into the follows: 0=<10%, 1=10-50%, 2=>50%. A positive stain was defined as 10% or more pathogenesis of pituitary tumors in patients with this disorder. immunoreactivity. Staining intensity was graded as 0 (negative), 1+ (weak), 2+ (moderate), or 3+ (strong). 428 CEAMCAM1 Is Expressed by Human Thyroid Carcinoma Cells Results: SDF-1 staining was observed in 12/16 papillary carcinomas, 1/2 follicular carcinomas, and 2/2 anaplastic carcinomas, while only 1/45 benign processes, a partially and Represents a Target for Vitamin D3 Therapy W Wei, W Liu, D Winer, AM Bamberger, S Ezzat, SL Asa. University Health Network, infarcted follicular adenoma, demonstrated immunoreactivity. 8/11 examples of University of Toronto, Toronto, ON, Canada; University Hospital, University of Hashimoto’s thyroiditis showed SDF-1 expression in atypical microscopic foci Hamburg, Hamburg, Germany; Mount Sinai Hospital, University of Toronto, Toronto, consisting of cells with incomplete morphologic features of papillary carcinoma and/ ON, Canada. or Hurthle cell-like features, in most cases accounting for 10-20% of follicular cells in Background: CEACAM1, formerly known as biliary glycoprotein, CD66a, C- the section. Statistical analysis of SDF-1 staining in malignant neoplasms of follicular CAM1 and pp120, is a member of the CEA immunoglobulin superfamily. CEACAM1 cell origin revealed sensitivity=75.0%, specificity=97.8%, PPV=93.8%, NPV= 89.8%. -

Research Article Identification of Immune-Related Genes in Sepsis Due to Community-Acquired Pneumonia
Hindawi Computational and Mathematical Methods in Medicine Volume 2021, Article ID 8020067, 11 pages https://doi.org/10.1155/2021/8020067 Research Article Identification of Immune-Related Genes in Sepsis due to Community-Acquired Pneumonia Yanyan Li,1 Jiqin Wang,1 Yuzhen Li,1 Chunyan Liu,1 Xia Gong,1 Yifei Zhuang,1 Liang Chen ,2 and Keyu Sun 1 1Department of Emergency, Minhang Hospital, Fudan University, No. 170 Xinsong Road, Minhang District, Shanghai 201199, China 2Department of Emergency Medicine, Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine, No. 1665 Kongjiang Road, Yangpu District, Shanghai 200092, China Correspondence should be addressed to Liang Chen; [email protected] and Keyu Sun; [email protected] Received 26 May 2021; Accepted 9 August 2021; Published 26 August 2021 Academic Editor: Tao Huang Copyright © 2021 Yanyan Li et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Background. Immunosuppression has a key function in sepsis pathogenesis, so it is of great significance to find immune-related markers for the treatment of sepsis. Methods. Datasets of community-acquired pneumonia (CAP) with sepsis from the ArrayExpress database were extracted. Differentially expressed genes (DEGs) between the CAP group and normal group by Limma package were performed. After calculation of immune score through the ESTIMATE algorithm, the DEGs were selected between the high immune score group and the low immune score group. Enrichment analysis of the intersected DEGs was conducted. Further, the protein-protein interaction (PPI) of the intersected DEGs was drawn by Metascape tools. -

A Neutrophil Activation Signature in Covid-19 Athanasios Didangelos
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 20 April 2020 doi:10.20944/preprints202004.0363.v1 A Neutrophil activation signature in Covid-19 Athanasios Didangelos (PhD) [email protected] University of Leicester, Mayer IgA Nephropathy Laboratory, University of Leicester, Leicester, LE1 7RH, United Kingdom Abstract Covid-19 is often related to hyperinflammation that drives lung or multi-organ damage and mortality. The immunopathological mechanisms that cause excessive inflammation following SARS-Cov-2 infection are under investigation while different approaches to limit hyperinflammation in affected patients are being proposed. Here, a computational network approach was used on recently available data to identify possible Covid-19 inflammatory mechanisms. First, network analysis of putative SARS-Cov-2 cellular receptors and their directly associated interacting proteins, led to the mining of a robust neutrophil-response signature and multiple relevant inflammatory response genes. Second, analysis of RNA-seq datasets of lung epithelial cells infected with SARS-Cov-2 found that infected cells specifically expressed neutrophil-attracting chemokines, further supporting the likely role of neutrophils in Covid-19 inflammation. The role of neutrophils in Covid-19 needs to be studied further. Different immunoregulatory molecules and pathways presented here (TNF Receptor, IL8, CXCR1, CXCR2, ADAM10, GPR84, MME-neprilysin, ANPEP, LAP3) are druggable and might be therapeutic targets in efforts to limit SARS-Cov-2 inflammation in severe clinical cases. Introduction New studies have highlighted that Covid-19 is often characterised by an extreme inflammatory response associated with lung and multi-organ injury and mortality and have suggested promising anti-inflammatory options (1). Other studies recommend caution with immunosuppression given that regulated inflammation is necessary for an effective anti-viral response (2). -

The Immuno‐Oncology Revolution Continues
CATALYST: The Immuno‐oncology Revolution Continues: A 3D View Chapter 3: Resistance or Nonresponse to Treatment Mario Sznol, MD Professor of Medicine (Medical Oncology) Co‐Director, Cancer Immunology Program at Yale Cancer Center New Haven, CT Disclosures Dr. Sznol has disclosed that he is a consultant for AbbVie, Allakos, Almac, AstraZeneca/Medimmune, Biodesix, Bristol‐ Myers Squibb, Genentech/Roche, Genmab, Hinge, Innate Pharma, Immunocore, Modulate Therapeutics, Molecular Partners, Newlink Genetics, Novartis, Torque, and Seattle Genetics. Dr. Sznol is also on scientific advisory boards for Adaptimmine, Lyciera, Omniox, Pieris, and Symphogen. This activity is supported by an educational grant from Bristol‐Myers Squibb. CME Objectives • Discuss the pathophysiology of adult malignancies with a focus on tumor immunosurveillance and immune evasion • Review significant advances and unmet medical needs associated with currently available immuno‐oncology therapies, including innate and adaptive resistance mechanisms (eg, T‐cell exhaustion) • Describe immune pathways that may be targeted to overcome immune‐evasion mechanisms and emerging clinical data on novel immuno‐oncology agents FDA‐Approved Cancer Immune Checkpoint Inhibitors Agent Target Cancer Indication(s) PD‐1/PD‐L1 Melanoma; NSCLC; metastatic SCLC; intermediate/advanced RCC; Nivolumab PD‐1 HCC; cHL; HNSCC; and urothelial and MSI‐H/dMMR cancers Melanoma; NSCLC; HNSCC; cHL; PMBCL; HCC; and urothelial, Pembrolizumab PD‐1 MSI‐H/dMMR, gastric, cervical, and Merkel cell cancers Atezolizumab