Insulin Sensitivity Is Retained in Mice with Endothelial Loss of Carcinoembryonic Antigen Cell Adhesion Molecule 1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

Prediction of Distant Metastasis by Using Reverse Transcriptase Polymerase Chain Reaction for Epithelial and Variant CD44 Mrna I
ORIGINAL ARTICLE Prediction of Distant Metastasis by Using Reverse Transcriptase–Polymerase Chain Reaction for Epithelial and Variant CD44 mRNA in the Peripheral Blood of Patients With Colorectal Cancer Shozo Yokoyama, MD; Hiroki Yamaue, MD Background: Reverse transcriptase–polymerase chain Main Outcome Measure: The clinical significance of reaction (RT-PCR) has been used to identify small num- RT-PCR for epithelial and variant CD44 mRNA in pe- bers of tumor cells. Molecular detection is thought to pro- ripheral blood. vide useful information for the clinical management of postoperative adjuvant therapy regimens. Results: During 3 years of follow-up, 2 patients whose peripheral blood had carcinoembryonic antigen and Objective: To use RT-PCR to identify messenger RNA CD44 variant mRNA also had distant metastases (lung (mRNA) coding for carcinoembryonic antigen, epithelial or spleen). Expression of epithelial and variant CD44 and variant CD44, and matrix metalloproteinase 7 in the mRNA in peripheral blood was more highly correlated portal venous and peripheral blood of patients with colo- with the clinical cancer stage than with expression rectal carcinoma to predict live or distant metastasis. of carcinoembryonic antigen and matrix metallopro- teinase 7. Design: Prospective consecutive series. Conclusions: Molecular detection of epithelial and Setting: University hospital. variant CD44 mRNA in the peripheral blood may help determine distant metastases in patients with colorectal Patients and Methods: Portal venous and peripheral carcinoma. Molecular detection in the peripheral blood blood samples were obtained from 22 patients with colo- at surgical treatment suggests that systemic hemato- rectal cancer during surgical manipulation. Using comple- genic tumor cell dissemination is an early event of dis- mentary DNA primers specific for carcinoembryonic tant metastasis. -

CD44 Predicts Early Recurrence in Pancreatic Cancer Patients Undergoing Radical Surgery
in vivo 32 : 1533-1540 (2018) doi:10.21873/invivo.11411 CD44 Predicts Early Recurrence in Pancreatic Cancer Patients Undergoing Radical Surgery CHIH-PO HSU 1* , LI-YU LEE 2* , JUN-TE HSU 1, YU-PAO HSU 1, YU-TUNG WU 1, SHANG-YU WANG 1, CHUN-NAN YEH 1, TSE-CHING CHEN 2 and TSANN-LONG HWANG 1 1Department of General Surgery, Chang Gung Memorial Hospital at Linkou, Chang Gung University College of Medicine, Taoyuan, Taiwan, R.O.C.; 2Department of Pathology, Chang Gung Memorial Hospital at Linkou, Chang Gung University College of Medicine, Taoyuan, Taiwan, R.O.C. Abstract. Background/Aim: Pancreatic ductal adeno- predicted ER. Conclusion: High CA19-9 levels, CD44 H- carcinoma (PDAC) is one of the most aggressive types of scores and poor differentiation are independent predictors digestive cancer. Recurrence within one year after surgery is for ER in PDAC patients undergoing radical resection. inevitable in most PDAC patients. Recently, cluster of Therefore, the determination of CD44 expression might help differentiation 44 (CD44) has been shown to be associated in identifying patients at a high risk of ER for more with tumor initiation, metastasis and prognosis. This study aggressive treatment after radical surgery. aimed to explore the correlation of CD44 expression with clinicopathological factors and the role of CD44 in Pancreatic ductal adenocarcinoma (PDAC) is the fourth most predicting early recurrence (ER) in PDAC patients after common cause of cancer-related death worldwide, with an radical surgery. Materials and Methods: PDAC patients who 8% 5-year survival rate for all stages of disease (1). underwent radical resection between January 1999 and Although various treatment modalities are available, only March 2015 were enrolled in this study. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

The Role of the Carcinoembryonic Antigen Receptor in Colorectal Cancer Progression
gra nte tive f I O o l n a c o n r l o u g o y J Bajenova et al., J Integr Oncol 2017, 6:2 Journal of Integrative Oncology DOI: 10.4172/2329-6771.1000192 ISSN: 2329-6771 Research Article Open Access The Role of the Carcinoembryonic Antigen Receptor in Colorectal Cancer Progression Olga Bajenova1,2, Elena Tolkunova3, Sergey Koshkin3, Sergey Malov1, Peter Thomas4, Alexey Tomilin3 and Stephen O’Brien1 1Theodosius Dobzhansky Center for Genome Bioinformatics at St. Petersburg State University, St. Petersburg, Russia 2Department of Genetics and Biotechnology, St. Petersburg State University, St. Petersburg, Russia 3Institute of Cytology, Russian Academy of Sciences, St. Petersburg, Russia 4Department of Surgery, Creighton University, Omaha, USA *Corresponding author: Olga Bajenova, Theodosius Dobzhansky Center for Genome Bioinformatics at St. Petersburg State University, 41-43 Sredniy Prospekt, St Petersburg, Russia, Tel: +7-812-363-6103; E-mail: [email protected] Received Date: March 25, 2017; Accepted Date: April 18, 2017; Published Date: April 28, 2017 Copyright: © 2017 Bajenova O, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Clinical and experimental data suggest that carcinoembryonic antigen (CEA, CD66e, CEACAM-5) plays a key role in the formation of hepatic metastasis from colorectal and other types of epithelial cancers. The molecular events involved in CEA-induced metastasis have yet to be defined. Our group first cloned the gene (CEAR) for CEA- binding protein from the surface of fixed liver macrophages, (Kupffer cells). -

Soluble Carcinoembryonic Antigen Activates Endothelial Cells and Tumor Angiogenesis
Published OnlineFirst October 11, 2013; DOI: 10.1158/0008-5472.CAN-13-0123 Cancer Microenvironment and Immunology Research Soluble Carcinoembryonic Antigen Activates Endothelial Cells and Tumor Angiogenesis Kira H. Bramswig1, Marina Poettler1, Matthias Unseld1, Friedrich Wrba2, Pavel Uhrin3, Wolfgang Zimmermann4, Christoph C. Zielinski1, and Gerald W. Prager1 Abstract Carcinoembryonic antigen (CEA, CD66e, CEACAM-5) is a cell-surface–bound glycoprotein overexpressed and released by many solid tumors that has an autocrine function in cancer cell survival and differentiation. Soluble CEA released by tumors is present in the circulation of patients with cancer, where it is used as a marker for cancer progression, but whether this form of CEA exerts any effects in the tumor microenvironment is unknown. Here, we present evidence that soluble CEA is sufficient to induce proangiogenic endothelial cell behaviors, including adhesion, spreading, proliferation, and migration in vitro and tumor microvascularization in vivo. CEA-induced activation of endothelial cells was dependent on integrin b-3 signals that activate the focal- adhesion kinase and c-Src kinase and their downstream MAP–ERK kinase/extracellular signal regulated kinase and phosphoinositide 3-kinase/Akt effector pathways. Notably, while interference with VEGF signaling had no effect on CEA-induced endothelial cell activation, downregulation with the CEA receptor in endothelial cells attenuated CEA-induced signaling and tumor angiogenesis. Corroborating these results clinically, we found -

Combination Immune Therapy
Combination Immune Therapy Translational Medicine Plenary SWOG April 17, 2017 San Francisco T‐cell Activation, Proliferation, and Function is Controlled by Multiple Agonist and Antagonist Signals 1. Co‐stimulation via CD28 ligation 2. CTLA‐4 ligation on activated T 3. T cell function in tissue is subject transduces T cell activating signals cells down‐regulates T cell to feedback inhibition responses T cell Functional Block T cell Activation T cell Activation T cell Proliferative Block T cell T cell APC T cell CTLA‐4 T cell CTLA‐4 TCR TCR PD‐1 PD‐1 PD‐1 IFN‐gamma TCR CD28 Cytokines TCR CD4 MHC MHC PD‐‐L1 MHC B7 MHC CD28 B7 PD‐‐L1 Tumor or Tumor or APC APC Immune cell B7 immune cell APC NK Presence of PD-L1 or TILs1 PPD-L1 /TIL+ PD-L1/TIL + + PD-L1 /TIL+ + PD-L1 /TIL PD-L1 /TIL D-L1 /TIL+ Schalper and Rimm, Yale University NSCLC PD-L1/TIL 45% 17% 26% 12% Type 1 Type 2 Type 3 Type 4 45% 41% 13% 1% Melanoma45% 17% 26% 12% Taube et al Type 1 Type 2 Type 3 Type 4 Tumor‐specific T cells are contained in the PD‐1+ TIL population and are functional after in vitro culture Spectrum of PD‐1/PD‐L1 Antagonist Activity Active • Melanoma Minimal to no activity • Renal cancer (clear cell and non‐clear cell) • Prostate cancer • NSCLC – adenocarcinoma and squamous cell • MMR+ (MSS) colon cancer • Small cell lung cancer • Myeloma • Head and neck cancer • Pancreatic cancer • Gastric and gastroesophageal junction • MMR‐repair deficient tumors (colon, cholangiocarcinoma) • Bladder • Triple negative breast cancer • Ovarian Major PD‐1/PD‐L1 antagonists • Hepatocellular -
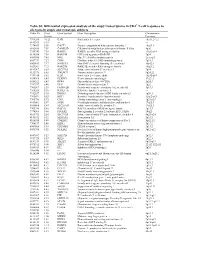
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3 -

Role of Liver ICAM‑1 in Metastasis (Review)
ONCOLOGY LETTERS 14: 3883-3892, 2017 Role of liver ICAM‑1 in metastasis (Review) AITOR BENEDICTO, IRENE ROMAYOR and BEATRIZ ARTETA Department of Cell Biology and Histology, School of Medicine and Nursing, University of The Basque Country, UPV/EHU, Leioa, E-48940 Vizcaya, Spain Received January 17, 2017; Accepted July 7, 2017 DOI: 10.3892/ol.2017.6700 Abstract. Intercellular adhesion molecule (ICAM)-1, is a frequently diagnosed cancer types and is the fourth leading transmembrane glycoprotein of the immunoglobulin (Ig)-like cause of cancer-related death (1). The spreading of cells from superfamily, consisting of five extracellular Ig‑like domains, a the primary lesion to a secondary organ and the subsequent transmembrane domain and a short cytoplasmic tail. ICAM-1 is development of distant metastases is a key factor that limits expressed in various cell types, including endothelial cells and patient survival rate. This remains one of the most complex leukocytes, and is involved in several physiological processes. issues faced in medicine (2). Furthermore, it has additionally been reported to be expressed The liver is the main target organ for metastatic CRC in various cancer cells, including melanoma, colorectal cancer cells and the second most commonly invaded organ, after and lymphoma. The majority of studies to date have focused the lymph nodes (3). In fact, 15-25% of CRC patients present on the expression of the ICAM‑1 on the surface of tumor cells, with synchronous hepatic metastases at the time of diagnosis, without research into ICAM‑1 expression at sites of metastasis. and a further 30% will later develop liver metastasis (4,5). -

Bioinformatics Method Identifies Potential Biomarkers of Dilated Cardiomyopathy in a Human Induced Pluripotent Stem Cell‑Derived Cardiomyocyte Model
EXPERIMENTAL AND THERAPEUTIC MEDICINE 14: 2771-2778, 2017 Bioinformatics method identifies potential biomarkers of dilated cardiomyopathy in a human induced pluripotent stem cell‑derived cardiomyocyte model YU ZHUANG1, YU-JIA GONG2, BEI-FEN ZHONG3, YI ZHOU1 and LI GONG4 1Department of Cardiovascular Surgery, Shanghai First People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080; 2Stomatology Faculty, School of Medicine, Nantong University, Nantong, Jiangsu 226000; 3Department of Urology, Shanghai First People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080; 4Department of Cardiothoracic Surgery, The Affiliated Huai'an Hospital of Xuzhou Medical University, Huai'an, Jiangsu 223002, P.R. China Received February 19, 2016; Accepted February 10, 2017 DOI: 10.3892/etm.2017.4850 Abstract. Dilated cardiomyopathy (DCM) is the most common crucial nodes in module 2, which were linked to each other. type of cardiomyopathy that account for the majority of heart In conclusion, several potential biomarkers for DCM were failure cases. The present study aimed to reveal the under- identified, such as MMP2, FLT1, CDH1, ITGB6, COL6A3, lying molecular mechanisms of DCM and provide potential COL6A1, LAMC2, PENK and APLNR. These genes may serve biomarkers for detection of this condition. The public dataset significant roles in DCM via involvement of various BPs, such of GSE35108 was downloaded, and 4 normal induced pluripo- as blood vessel and vasculature development and cell adhe- tent stem cell (iPSC)-derived cardiomyocytes (N samples) and sion, and the ECM-receptor interaction pathway. 4 DCM iPSC-derived cardiomyocytes (DCM samples) were utilized. Raw data were preprocessed, followed by identifica- Introduction tion of differentially expressed genes (DEGs) between N and DCM samples. -

The Immunomodulatory CEA Cell Adhesion Molecule 6 (CEACAM6/Cd66c) Is a Candidate Receptor for the Influenza a Virus
bioRxiv preprint doi: https://doi.org/10.1101/104026; this version posted January 30, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 The immunomodulatory CEA cell adhesion molecule 6 (CEACAM6/CD66c) is a 2 candidate receptor for the influenza A virus 3 Shah Kamranur Rahmana *, Mairaj Ahmed Ansarib, Pratibha Gaurc, Imtiyaz Ahmada, 4 Chandrani Chakravartya,d, Dileep Kumar Vermaa, Sanjay Chhibbere, Naila Nehalf, 5 Shanmugaapriya Sellathanbyd, Dagmar Wirthc, Gulam Warisb and Sunil K. Lala,g # 6 7 Virology Group, International Centre for Genetic Engineering & Biotechnology, New Delhi, 8 Indiaa. 9 Department of Microbiology and Immunology, H. M. Bligh Cancer Research Laboratories, 10 Rosalind Franklin University of Medicine and Science, Chicago Medical School, North 11 Chicago, Illinois, USAb. 12 Helmholtz Centre for Infection Research, Braunschweig, Germanyc. 13 Department of Biomedical Science, Bharathidasan University, Trichy, Indiad. 14 Microbiology Department, Panjab University, Chandigarh, Indiae. 15 Career Institute of Medical & Dental Sciences and Hospital, Lucknow, Indiaf. 16 School of Science, Monash University, Selangor DE, Malaysiag. 17 18 Running Head: Protein receptor for Influenza A Virus 19 20 # Corresponding author: Professor of Microbiology, School of Science, Monash University, 21 47500 Bandar Sunway, Selangor DE, Malaysia. 22 Email: [email protected]; Telephone: (+603) 551 59606 23 24 * Current address: Department of Pathogen Molecular Biology, London School of Hygiene & 25 Tropical Medicine, Keppel Street, London WC1E 7HT, United Kingdom. 26 1 bioRxiv preprint doi: https://doi.org/10.1101/104026; this version posted January 30, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Receptor for Mouse Hepatitis Virus Is a Member of the Carcinoembryonic Antigen Family of Glycoproteins
Proc. Natl. Acad. Sci. USA Vol. 88, pp. 5533-5536, July 1991 Medical Sciences Receptor for mouse hepatitis virus is a member of the carcinoembryonic antigen family of glycoproteins (coronavirus/immunoglobulin superfamily/host resistance to virus/virus tissue tropism/virus receptor) RICHARD K. WILLIAMS, GUI-SEN JIANG, AND KATHRYN V. HOLMES* Department of Pathology, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, Bethesda, MD 20814 Communicated by Dorothy M. Horstmann, April 1, 1991 (receivedfor review February 28, 1991) ABSTRACT The receptor for mouse hepatitis virus binds neither MHV-A59 virus nor anti-receptor mAb CC1, (MHV), a murine coronavirus, is a 110- to 120-kDa glycopro- we postulated that it has a mutation affecting the domain of tein on intestinal brush border membranes and hepatocyte the glycoprotein that normally would bind MHV and mAb membranes. The N-terminal 25-amino acid sequence of immu- CC1 (4). In this communication we demonstrate that the noaffinity-purified MHV receptor was identical to the pre- MHV receptor glycoprotein of BALB/c and Swiss Webster dicted mature N termini of two mouse genes related to human mice and its SJL/J homolog are members of the carcinoem- carcinoembryonic antigen (CEA) and was strongly homologous bryonic antigen (CEA) family of glycoproteins in the immu- to the N termini of members of the CEA family in humans and noglobulin superfamily. rats. Polyclonal antibodies to human CEA recognized the immunoaffinity-purified MHV receptor and the MHV receptor in liver membranes and intestinal brush border membranes MATERIALS AND METHODS from MHV-susceptible mouse strains. In membranes from Tissue Preparations and Tissue-Binding Assay.