MOP): Clinical and Data Management for Using Biomarkers to Predict TB Treatment Duration (Predict TB Trial
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

World Bank Document
CONFORMED COPY LOAN NUMBER 7909-CN Public Disclosure Authorized Project Agreement Public Disclosure Authorized (Henan Ecological Livestock Project) between INTERNATIONAL BANK FOR RECONSTRUCTION AND DEVELOPMENT Public Disclosure Authorized and HENAN PROVINCE Dated July 26, 2010 Public Disclosure Authorized PROJECT AGREEMENT AGREEMENT dated July 26, 2010, entered into between INTERNATIONAL BANK FOR RECONSTRUCTION AND DEVELOPMENT (the “Bank”) and HENAN PROVINCE (“Henan” or the “Project Implementing Entity”) (“Project Agreement”) in connection with the Loan Agreement of same date between PEOPLE’S REPUBLIC OF CHINA (“Borrower”) and the Bank (“Loan Agreement”) for the Henan Ecological Livestock Project (the “Project”). The Bank and Henan hereby agree as follows: ARTICLE I – GENERAL CONDITIONS; DEFINITIONS 1.01. The General Conditions as defined in the Appendix to the Loan Agreement constitute an integral part of this Agreement. 1.02. Unless the context requires otherwise, the capitalized terms used in the Project Agreement have the meanings ascribed to them in the Loan Agreement or the General Conditions. ARTICLE II – PROJECT 2.01. Henan declares its commitment to the objective of the Project. To this end, Henan shall: (a) carry out the Project in accordance with the provisions of Article V of the General Conditions; and (b) provide promptly as needed, the funds, facilities, services and other resources required for the Project. 2.02. Without limitation upon the provisions of Section 2.01 of this Agreement, and except as the Bank and Henan shall otherwise agree, Henan shall carry out the Project in accordance with the provisions of the Schedule to this Agreement. ARTICLE III – REPRESENTATIVE; ADDRESSES 3.01. -

From the Land of Henan, for the People of China
From the land of Henan, for the people of China. 2015 ANNUAL REPORT CONTENTS CORPORATE INFORMATION 2 DIRECTORS’ REPORT 68 CORPORATE PROFILE 4 INDEPENDENT AUDITOR’S REPORT 80 CHAIRMAN’S STATEMENT 9 CONSOLIDATED INCOME STATEMENT 82 FINANCIAL HIGHLIGHTS 12 CONSOLIDATED STATEMENT OF COMPREHENSIVE INCOME 83 MANAGEMENT DISCUSSION AND ANALYSIS 13 CONSOLIDATED STATEMENT OF FINANCIAL POSITION 84 I. FINANCIAL REVIEW 14 CONSOLIDATED STATEMENT OF CHANGES IN EQUITY 86 II. REVIEW OF OPERATIONS 18 CONSOLIDATED CASH FLOW STATEMENT 88 III. BUSINESS OUTLOOK 38 NOTES TO THE FINANCIAL STATEMENTS 91 INVESTOR RELATIONS REPORT 43 SUMMARY OF FINANCIAL INFORMATION 183 ENVIRONMENTAL, SOCIAL AND GOVERNANCE REPORT 47 CORPORATE GOVERNANCE REPORT 53 PROFILE OF DIRECTORS AND SENIOR MANAGEMENT 64 Xuchang Yanling Eco-City Corporate Information BOARD OF DIRECTORS COMPANY SECRETARY Executive Directors Mr. Kwok Pak Shing Mr. Wu Po Sum (Chairman) Ms. Yan Yingchun REGISTERED OFFICE Cricket Square Non-executive Directors Hutchins Drive Mr. Lucas Ignatius Loh Jen Yuh (Vice-Chairman) P.O. Box 2681 Mr. Puah Tze Shyang (appointed on 1 April 2015) Grand Cayman Mr. Leow Juan Thong Jason (resigned on 1 April 2015) KY1-1111 Ms. Wu Wallis (alias Li Hua) Cayman Islands Independent Non-executive Directors PRINCIPAL PLACE OF BUSINESS IN THE PRC Mr. Cheung Shek Lun Block E, Jianye Office Building Mr. Xin Luo Lin Nongye East Road, Zhengzhou City Mr. Muk Kin Yau Henan Province, the PRC BOARD COMMITTEES PRINCIPAL PLACE OF BUSINESS IN HONG KONG Audit Committee Room 7701B–7702A Mr. Cheung Shek Lun (Chairman) 77th Floor, International Commerce Centre Mr. Xin Luo Lin 1 Austin Road West Mr. -

CHINA VANKE CO., LTD.* 萬科企業股份有限公司 (A Joint Stock Company Incorporated in the People’S Republic of China with Limited Liability) (Stock Code: 2202)
Hong Kong Exchanges and Clearing Limited and The Stock Exchange of Hong Kong Limited take no responsibility for the contents of this announcement, make no representation as to its accuracy or completeness and expressly disclaim any liability whatsoever for any loss howsoever arising from or in reliance upon the whole or any part of the contents of this announcement. CHINA VANKE CO., LTD.* 萬科企業股份有限公司 (A joint stock company incorporated in the People’s Republic of China with limited liability) (Stock Code: 2202) 2019 ANNUAL RESULTS ANNOUNCEMENT The board of directors (the “Board”) of China Vanke Co., Ltd.* (the “Company”) is pleased to announce the audited results of the Company and its subsidiaries for the year ended 31 December 2019. This announcement, containing the full text of the 2019 Annual Report of the Company, complies with the relevant requirements of the Rules Governing the Listing of Securities on The Stock Exchange of Hong Kong Limited in relation to information to accompany preliminary announcement of annual results. Printed version of the Company’s 2019 Annual Report will be delivered to the H-Share Holders of the Company and available for viewing on the websites of The Stock Exchange of Hong Kong Limited (www.hkexnews.hk) and of the Company (www.vanke.com) in April 2020. Both the Chinese and English versions of this results announcement are available on the websites of the Company (www.vanke.com) and The Stock Exchange of Hong Kong Limited (www.hkexnews.hk). In the event of any discrepancies in interpretations between the English version and Chinese version, the Chinese version shall prevail, except for the financial report prepared in accordance with International Financial Reporting Standards, of which the English version shall prevail. -

Announcement of Interim Results for the Six Months Ended 30 June 2021
Hong Kong Exchanges and Clearing Limited and The Stock Exchange of Hong Kong Limited take no responsibility for the contents of this announcement, make no representation as to its accuracy or completeness and expressly disclaim any liability whatsoever for any loss howsoever arising from or in reliance upon the whole or any part of the contents of this announcement. (Stock Code: 0832) ANNOUNCEMENT OF INTERIM RESULTS FOR THE SIX MONTHS ENDED 30 JUNE 2021 FINANCIAL HIGHLIGHTS • Revenue for the six months ended 30 June 2021 amounted to RMB20,357 million, an increase of 56.4% compared with the corresponding period in 2020. • Gross profit margin for the period was 17.9%, a decrease of 5.8 percentage points compared with 23.7% for the corresponding period in 2020. • Profit attributable to equity shareholders of the Company for the period amounted to RMB729 million, an increase of 0.3% compared with the corresponding period in 2020. • Profit for the period was RMB1,025 million, representing an increase of 30.4% compared with the corresponding period in 2020. • Net profit margin for the period was 5.0%, a decrease of 1.0 percentage point compared with 6.0% for the corresponding period in 2020. • Basic earnings per share for the period was RMB25.63 cents, a decrease of 3.0% compared with the corresponding period in 2020. • Declaration of an interim dividend of HK14.75 cents per share for the six months ended 30 June 2021. 1 INTERIM RESULTS The board (the “Board”) of directors (the “Directors” and each a “Director”) of Central China Real Estate -

十六shí Liù Sixteen / 16 二八èr Bā 16 / Sixteen 和hé Old Variant of 和/ [He2
十六 shí liù sixteen / 16 二八 èr bā 16 / sixteen 和 hé old variant of 和 / [he2] / harmonious 子 zǐ son / child / seed / egg / small thing / 1st earthly branch: 11 p.m.-1 a.m., midnight, 11th solar month (7th December to 5th January), year of the Rat / Viscount, fourth of five orders of nobility 亓 / 等 / 爵 / 位 / [wu3 deng3 jue2 wei4] 动 dòng to use / to act / to move / to change / abbr. for 動 / 詞 / |动 / 词 / [dong4 ci2], verb 公 gōng public / collectively owned / common / international (e.g. high seas, metric system, calendar) / make public / fair / just / Duke, highest of five orders of nobility 亓 / 等 / 爵 / 位 / [wu3 deng3 jue2 wei4] / honorable (gentlemen) / father-in 两 liǎng two / both / some / a few / tael, unit of weight equal to 50 grams (modern) or 1&frasl / 16 of a catty 斤 / [jin1] (old) 化 huà to make into / to change into / -ization / to ... -ize / to transform / abbr. for 化 / 學 / |化 / 学 / [hua4 xue2] 位 wèi position / location / place / seat / classifier for people (honorific) / classifier for binary bits (e.g. 十 / 六 / 位 / 16-bit or 2 bytes) 乎 hū (classical particle similar to 於 / |于 / [yu2]) in / at / from / because / than / (classical final particle similar to 嗎 / |吗 / [ma5], 吧 / [ba5], 呢 / [ne5], expressing question, doubt or astonishment) 男 nán male / Baron, lowest of five orders of nobility 亓 / 等 / 爵 / 位 / [wu3 deng3 jue2 wei4] / CL:個 / |个 / [ge4] 弟 tì variant of 悌 / [ti4] 伯 bó father's elder brother / senior / paternal elder uncle / eldest of brothers / respectful form of address / Count, third of five orders of nobility 亓 / 等 / 爵 / 位 / [wu3 deng3 jue2 wei4] 呼 hū variant of 呼 / [hu1] / to shout / to call out 郑 Zhèng Zheng state during the Warring States period / surname Zheng / abbr. -

Bank of Zhengzhou Co., Ltd.* 鄭 州 銀 行 股 份 有 限 公 司*
The Stock Exchange of Hong Kong Limited and Securities and Futures Commission take no responsibility for the contents of this Application Proof, make no representation as to its accuracy or completeness and expressly disclaim any liability whatsoever for any loss howsoever arising from or in reliance upon the whole or any part of the contents of this Application Proof. Application Proof of BANK OF ZHENGZHOU CO., LTD.* 鄭州銀行股份有限公司* (A joint stock company incorporated in the People’s Republic of China with limited liability) WARNING The publication of this Application Proof is required by The Stock Exchange of Hong Kong Limited (the “Stock Exchange”) / the Securities and Futures Commission (the “Commission”) solely for the purpose of providing information to the public in Hong Kong. This Application Proof is in draft form. The information contained in it is incomplete and is subject to change which can be material. By viewing this document, you acknowledge, accept and agree with Bank of Zhengzhou Co., Ltd. ( the “Bank”), its sponsor, advisers or members of the underwriting syndicate that: (a) this document is only for the purpose of providing information about the Bank to the public in Hong Kong and not for any other purposes. No investment decision should be based on the information contained in this document; (b) the publication of this document or supplemental, revised or replacement pages on the Stock Exchange’s website does not give rise to any obligation of the Bank, its sponsors, advisers or members of the underwriting syndicate to proceed with an offering in Hong Kong or any other jurisdiction. -

Academic Year 2011-2012 STUDY on the ROLE of URBAN and PERI
Academic year 2011-2012 STUDY ON THE ROLE OF URBAN AND PERI-URBAN AGRICULTURE: A CASE OF ZHENGZHOU, CHINA Guo, Liangyan Promotor: Prof. Dr. Wolfgang Bokelmann Co-promotor: Prof. Francesco Di Iacovo Thesis submitted in partial fulfilment of the requirements for the joint academic degree of International Master of Science in Rural Development from Ghent University (Belgium), Agrocampus Ouest (France), Humboldt University of Berlin (Germany), Slovak University of Agriculture in Nitra (Slovakia) and University of Pisa (Italy) in collaboration with Wageningen University (The Netherlands). This thesis was elaborated and defended at Humboldt university of Berlin within the framework of the European Erasmus Mundus Programme “Erasmus Mundus International Master of Science in Rural Development “(Course N° 2010-0114 – R 04-018/001) Certification This is an unpublished M.Sc. thesis and is not prepared for further distribution. The author and the promoter give the permission to use this thesis for consultation and to copy parts of it for personal use. Every other use is subject to the copyright laws; more specifically the source must be extensively specified when using results from this thesis. The Promoter(s): Wolfgang Bolelmann The Author: Guo, Liangyan Thesis online access release I hereby authorize the IMRD secretariat to make this thesis available on line on the IMRD website The Author: Guo, Liangyan I Acknowledgements Firstly, I would like to thank my prompter Prof. Dr. Wolfgang Bolelmann, in his lecture it was the first time I learn about urban agriculture, and then gave me useful suggestion on the research questions. Also many thanks to all those people contributed to this thesis, my local friend who accompanied me in the field work and provided me lot of useful information and thanks to household in the interviews. -

Polycyclic Aromatic Hydrocarbon Distribution in Soil and Their Ecological Risk Evaluation in Zhengzhou, China
Nature Environment and Pollution Technology p-ISSN: 0972-6268 Vol. 16 No. 1 pp. 315-319 2017 An International Quarterly Scientific Journal e-ISSN: 2395-3454 Original Research Paper Open Access Polycyclic Aromatic Hydrocarbon Distribution in Soil and Their Ecological Risk Evaluation in Zhengzhou, China Shubo Cheng*† and Yongmei Zhang*(**) *Henan Safety and Emergency Research Center, Henan Polytechnic University, Jiaozuo, Henan, 454000, China **Emergency Management School, Henan Polytechnic University, Jiaozuo, Henan, 454000, China †Corresponding author: Shubo Cheng ABSTRACT Nat. Env. & Poll. Tech. Website: www.neptjournal.com Polycyclic aromatic hydrocarbon (PAH), as a primary organic pollutant in soil, is highly stable in soil and Received: 14-12-2016 does not degrade easily. Thus, it accumulates and may penetrate deeper soil, which will cause long- Accepted: 17-01-2017 term pollution of soil and underground water. To study PAH concentration distribution in soil in Zhengzhou, China, ecological risk evaluation of PAHs in soil was implemented. In this study, 30 soil samples from Key Words: different places in Zhengzhou were collected in August 2015. Concentration levels and distribution PAHs distribution characteristics of PAHs in the soil samples were analysed, and ecological risk evaluation of PAH Soil pollution in soil was conducted. Results indicated that among the 30 soil samples collected, the content Ecological risk evaluation scope of 16 PAHs under priority control of the US Environmental Protection Agency was 436.37- 4,784.13 µg.kg-1 with a mean value of 3,096.71 µg.kg-1. Detection rates of monomers were high, and not all benzo[k]fluoranthene (BkF) and dibenzo[a,h]anthracene were detected. -
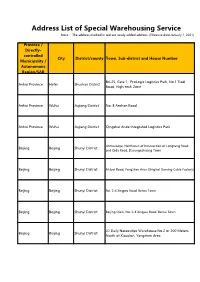
Address List of Special Warehousing Service Note: the Address Marked in Red Are Newly Added Address
Address List of Special Warehousing Service Note: The address marked in red are newly added address. (Effective date:January 1, 2021) Province / Directly- controlled City District/county Town, Sub-district and House Number Municipality / Autonomous Region/SAR B4-25, Gate 1, ProLogis Logistics Park, No.1 Tiedi Anhui Province Hefei Shushan District Road, High-tech Zone Anhui Province Wuhu Jiujiang District No. 8 Anshan Road Anhui Province Wuhu Jiujiang District Qingshui Ande Integrated Logistics Park Jinmucaoye, Northwest of Intersection of Longtang Road Beijing Beijing Shunyi District and Qida Road, Dasungezhuang Town Beijing Beijing Shunyi District Mubei Road, Yangzhen Area (Original Daming Cable Factory) Beijing Beijing Shunyi District No. 2-4 Xingwu Road, Beiwu Town Beijing Beijing Shunyi District Beijing Qixin, No. 2-4 Xingwu Road, Beiwu Town JD Daily Necessities Warehouse No.2 or 200 Meters Beijing Beijing Shunyi District North of Xiaodian, Yangzhen Area Daming Cable Factory or JD Warehouse No.3 , Beijing Beijing Shunyi District Xiaodian, Yangzhen Area 50 Meters North of Beijing Warehouse, Shaziying Village Bus Sichuan Province Beijing Shunyi District Stop, Yangzhen Beijing Beijing Shunyi District 200 Meters North of Xiaodian Middle School, Yangzhen Beijing Beijing Shunyi District No. 33 Zhangliang Road Beijing Beijing Shunyi District Andahui Warehouse, No. 33 Zhangliang Road No.1 Warehouse, Building Material Warehouse A, Beijing Beijing Beijing Shunyi District Department Store, No. 33 Zhangliang Road SF Cold Chain, Rongshang 3rd Road, Xingmao 2nd Beijing Beijing Tongzhou District Street, Majuqiao Sichuan Province Beijing Chaoyang District No.1-1, Kanghua Road, Laojuntang Village 5/F, Tiejian Warehouse #4, CR Logistics Base, No. 11 Beijing Beijing Tongzhou District Xingmao 1st Street Beijing Zhonghan United Lifting Machinery Co.,Ltd., Beijing Beijing Shunyi District No. -

CCICED Policy Research Report on Environment and Development 2018
CCICED Policy Research Report on Environment and Development 2018 Advisors Arthur HANSON(Canada), LIU Shijin Expert Board GUO Jing, FANG Li, ZHANG Yongsheng, LI Yonghong, ZHANG Jianyu, Knut ALFSEN(Norway), Dimitri De BOER(Netherlands), QIN Hu Editorial Board WANG Yong, ZHANG Huiyong, WU Jianmin Lucie McNEILL(Canada), DAI Yichun(Canada), ZHANG Jianzhi, LI Gongtao, LI Ying, ZHANG Min, LIU Qi, FEI Chengbo, JING Fang YAO Ying, LI Yutong A GREEN NEW ERA A GREEN FOR INNOVATION 01 Environment and Development Secretariat China Council for International Cooperation on Edited by 2CHINA COUNCIL FOR INTERNATIONAL COOPERATION ON ENVIRONMENT AND DEVELOPMENT POLICY RESEARCH REPORT ON ENVIRONMENT AND DEVELOPMENT 8 China Environment Publishing Group·Beijing 图书在版编目(CIP)数据 中国环境与发展国际合作委员会环境与发展政策研究 报 告 .2018, 创 新 引 领 绿 色 新 时 代 = CHINA COUNCIL FOR INTERNATIONAL COOPERATION ON ENVIRONMENT AND DEVELOPMENT POLICY RESEARCH REPORT ON ENVIRONMENT AND DEVELOPMENT 2018:INNOVATION FOR A GREEN NEW ERA: 英文 / 中国环境与发展 国际合作委员会秘书处编 . -- 北京 : 中国环境出版集团 , 2019.6 ISBN 978-7-5111-3974-0 Ⅰ . ①中… Ⅱ . ①中… Ⅲ . ①环境保护-研究报告-中国- 2018 -英文 Ⅳ . ① X-12 中国版本图书馆 CIP 数据核字 (2019) 第 085328 号 出 版 人 武德凯 责任编辑 黄 颖 张秋辰 责任校对 任 丽 装帧设计 宋 瑞 出版发行 中国环境出版集团 (100062 北京市东城区广渠门内大街 16 号) 网 址:http://www.cesp.com.cn 电子邮箱:[email protected] 联系电话:010-67112765(编辑管理部) 发行热线:010-67125803,010-67113405(传真) 印 刷 北京建宏印刷有限公司 经 销 各地新华书店 版 次 2019 年 6 月第 1 版 印 次 2019 年 6 月第 1 次印刷 开 本 787×960 1/16 印 张 10 字 数 165 千字 定 价 60.00 元 【版权所有。未经许可,请勿翻印、转载,违者必究。】 如有缺页、破损、倒装等印装质量问题,请寄回本社更换 Note on this volume China Council for International Cooperation on Environment and Development (CCICED) held the 2018 Annual General Meeting (AGM) with the theme of "Innovation for a Green New Era" from November 1st to 3rd. -

Annual Report
Important Notice: 1. The Board, the Supervisory Committee and the Directors, members of the Supervisory Committee and senior management of the Company warrant that in respect of the information contained in 2020 Annual Report (the “Report”, or “Annual Report”), there are no misrepresentations, misleading statements or material omission, and individually and collectively accept full responsibility for the authenticity, accuracy and completeness of the information contained in the Report. 2. The Report has been approved by the sixth meeting of the 19th session of the Board (the “Meeting”) convened on 30 March 2021. Mr. XIN Jie and Mr. TANG Shaojie, both being Non-executive Directors, did not attend the Meeting due to business engagement, and had authorised Mr. LI Qiangqiang, also a Non-executive Director, to attend the Meeting and executed voting rights on their behalf. All other Directors attended the Meeting in person. 3. The Company’s proposal on dividend distribution for the year of 2020: Based on the total share capital on the equity registration date when dividends are paid, the total amount of cash dividends proposed for distribution for 2020 will be RMB14,522,165,251.25 (inclusive of tax), accounting for 34.98% of the net profit attributable to equity shareholders of the Company for 2020, without any bonus shares or transfer of equity reserve to the share capital. Based on the Company’s total number of 11,617,732,201 shares at the end of 2020, a cash dividend of RMB12.5 (inclusive of tax) will be distributed for each 10 shares. If any circumstances, such as issuance of new shares, share repurchase or conversion of any convertible bonds into share capital before the record date for dividend distribution, results in the changes in our total number of shares on record date for dividend distribution, dividend per share shall be adjusted accordingly on the premise that the total dividends amount remains unchanged. -

The Journal of American Science ISSN 1545-1003 Volume 5 - Number 2, March 20, 2009
The Journal of American Science ISSN 1545-1003 Volume 5 - Number 2, March 20, 2009 © 2009 Marsland Press, the United States Journal of American Science ISSN 1545-1003 Journal of American Science The Journal of American Science is an international journal with a purpose to enhance our natural and scientific knowledge dissemination in the world under the free publication principle. Any valuable paper that describes natural phenomena and existence or any report that conveys scientific research and pursuit are welcome, including both natural and social sciences. Papers submitted could be reviews, objective descriptions, research reports, opinions/debates, news, letters, and other types of writings that are nature and science related. The journal is calling for papers and seeking co-operators and editors as well. Editor-in-Chief: Hongbao Ma Associate Editors-in-Chief: Shen Cherng, Jingjing Z Edmondson, Qiang Fu, Yongsheng Ma Editors: George Chen, Mark Hansen, Mary Herbert, Deng-Nan Horng, Wayne Jiang, Mark Lindley, Margaret D. Ma, Mike Ma, Da Ouyang, Xiaofeng Ren, Ajaya Kumar Sahoo, Shufang Shi, Tracy X Qiao, Pankaj Sah, George Warren, Qing Xia, Yonggang Xie, Lijian Yang, Jenny Young, Tina Zhang, Ruanbao Zhou, Yi Zhu Web Design: Jenny Young Introductions to Authors 1. General Information Reference Examples: (1) Goals: As an international journal published both in print and on Journal Article: Hacker J, Hentschel U, Dobrindt U. Prokaryotic internet, the Journal of American Science is dedicated to the chromosomes and disease. Science 2003;301(34):790-3. dissemination of fundamental knowledge in all areas of nature and Book: Berkowitz BA, Katzung BG. Basic and clinical evaluation of science.