Structure and Performance of Zeolite Supported Pd for Complete Methane Oxidation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Disability Classification System
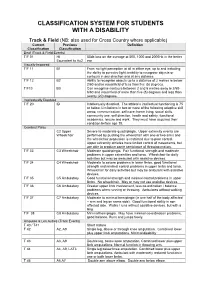
CLASSIFICATION SYSTEM FOR STUDENTS WITH A DISABILITY Track & Field (NB: also used for Cross Country where applicable) Current Previous Definition Classification Classification Deaf (Track & Field Events) T/F 01 HI 55db loss on the average at 500, 1000 and 2000Hz in the better Equivalent to Au2 ear Visually Impaired T/F 11 B1 From no light perception at all in either eye, up to and including the ability to perceive light; inability to recognise objects or contours in any direction and at any distance. T/F 12 B2 Ability to recognise objects up to a distance of 2 metres ie below 2/60 and/or visual field of less than five (5) degrees. T/F13 B3 Can recognise contours between 2 and 6 metres away ie 2/60- 6/60 and visual field of more than five (5) degrees and less than twenty (20) degrees. Intellectually Disabled T/F 20 ID Intellectually disabled. The athlete’s intellectual functioning is 75 or below. Limitations in two or more of the following adaptive skill areas; communication, self-care; home living, social skills, community use, self direction, health and safety, functional academics, leisure and work. They must have acquired their condition before age 18. Cerebral Palsy C2 Upper Severe to moderate quadriplegia. Upper extremity events are Wheelchair performed by pushing the wheelchair with one or two arms and the wheelchair propulsion is restricted due to poor control. Upper extremity athletes have limited control of movements, but are able to produce some semblance of throwing motion. T/F 33 C3 Wheelchair Moderate quadriplegia. Fair functional strength and moderate problems in upper extremities and torso. -

Framework for In-Field Analyses of Performance and Sub-Technique Selection in Standing Para Cross-Country Skiers
sensors Article Framework for In-Field Analyses of Performance and Sub-Technique Selection in Standing Para Cross-Country Skiers Camilla H. Carlsen 1,*, Julia Kathrin Baumgart 1, Jan Kocbach 1,2, Pål Haugnes 1 , Evy M. B. Paulussen 1,3 and Øyvind Sandbakk 1 1 Centre for Elite Sports Research, Department of Neuromedicine and Movement Science, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, 7491 Trondheim, Norway; [email protected] (J.K.B.); [email protected] (J.K.); [email protected] (P.H.); [email protected] (E.M.B.P.); [email protected] (Ø.S.) 2 NORCE Norwegian Research Centre AS, 5008 Bergen, Norway 3 Faculty of Health, Medicine & Life Sciences, Maastricht University, 6200 MD Maastricht, The Netherlands * Correspondence: [email protected]; Tel.: +47-452-40-788 Abstract: Our aims were to evaluate the feasibility of a framework based on micro-sensor technology for in-field analyses of performance and sub-technique selection in Para cross-country (XC) skiing by using it to compare these parameters between elite standing Para (two men; one woman) and able- bodied (AB) (three men; four women) XC skiers during a classical skiing race. The data from a global navigation satellite system and inertial measurement unit were integrated to compare time loss and selected sub-techniques as a function of speed. Compared to male/female AB skiers, male/female Para skiers displayed 19/14% slower average speed with the largest time loss (65 ± 36/35 ± 6 s/lap) Citation: Carlsen, C.H.; Kathrin found in uphill terrain. -

Rev Bras Cineantropomhum
Rev Bras Cineantropom Hum original article DOI: http://dx.doi.org/10.5007/1980-0037.2017v19n2p196 Sport classification for athletes with visual impairment and its relation with swimming performance Classificação esportiva para atletas com deficiência visual e sua relação com o desempenho na natação Elaine Cappellazzo Souto1,2 Leonardo dos Santos Oliveira1 Claudemir da Silva Santos2 Márcia Greguol1 Abstract – The medical classification (MC) adopted for swimmers with vision visual impairment (VI) does not clearly elucidate the influence of vision loss on performance. In a documentary research, the final time in the 50-, 100- and 400-m freestyle events and MC (S11, S12 and S13) of national (n = 40) and international (n = 72) elite swimmers was analyzed. The analysis was performed using the Kruskal-Wallis test and Spearman’s correlation with 95% confidence (P < 0.05) and Cohen’s d was calculated. There was a large effect of MC on the final time in the 50-m (P = 0.034, d = 1.55) for national ath- letes and in the 50-m (P = 0.001, d = 2.64), 100-m (P = 0.001, d = 3.01) and 400-m (P = 0.001, d = 2.88) for international athletes. S12 and S13 classes were faster compared to S11 class for all international events, but only in the 50-m for the national event (P < 0.05). It was found a strong negative relationship between the final time and MC for international athletes (Spearman’s Rho ≥ 0.78). There was a significant influence of MC on the performance of swimmers in freestyle races, especially in international swimmers. -

National Classification? 13
NATIONAL CL ASSIFICATION INFORMATION FOR MULTI CLASS SWIMMERS Version 1.2 2019 PRINCIPAL PARTNER MAJOR PARTNERS CLASSIFICATION PARTNERS Version 1.2 2019 National Swimming Classification Information for Multi Class Swimmers 1 CONTENTS TERMINOLOGY 3 WHAT IS CLASSIFICATION? 4 WHAT IS THE CLASSIFICATION PATHWAY? 4 WHAT ARE THE ELIGIBLE IMPAIRMENTS? 5 CLASSIFICATION SYSTEMS 6 CLASSIFICATION SYSTEM PARTNERS 6 WHAT IS A SPORT CLASS? 7 HOW IS A SPORT CLASS ALLOCATED TO AN ATHLETE? 7 WHAT ARE THE SPORT CLASSES IN MULTI CLASS SWIMMING? 8 SPORT CLASS STATUS 11 CODES OF EXCEPTION 12 HOW DO I CHECK MY NATIONAL CLASSIFICATION? 13 HOW DO I GET A NATIONAL CLASSIFICATION? 13 MORE INFORMATION 14 CONTACT INFORMATION 16 Version 1.2 2019 National Swimming Classification Information for Multi Class Swimmers 2 TERMINOLOGY Assessment Specific clinical procedure conducted during athlete evaluation processes ATG Australian Transplant Games SIA Sport Inclusion Australia BME Benchmark Event CISD The International Committee of Sports for the Deaf Classification Refers to the system of grouping athletes based on impact of impairment Classification Organisations with a responsibility for administering the swimming classification systems in System Partners Australia Deaflympian Representative at Deaflympic Games DPE Daily Performance Environment DSA Deaf Sports Australia Eligibility Criteria Requirements under which athletes are evaluated for a Sport Class Evaluation Process of determining if an athlete meets eligibility criteria for a Sport Class HI Hearing Impairment ICDS International Committee of Sports for the Deaf II Intellectual Impairment Inas International Federation for Sport for Para-athletes with an Intellectual Disability General term that refers to strategic initiatives that address engagement of targeted population Inclusion groups that typically face disadvantage, including people with disability. -

United States Olympic Committee and U.S. Department of Veterans Affairs
SELECTION STANDARDS United States Olympic Committee and U.S. Department of Veterans Affairs Veteran Monthly Assistance Allowance Program The U.S. Olympic Committee supports Paralympic-eligible military veterans in their efforts to represent the USA at the Paralympic Games and other international sport competitions. Veterans who demonstrate exceptional sport skills and the commitment necessary to pursue elite-level competition are given guidance on securing the training, support, and coaching needed to qualify for Team USA and achieve their Paralympic dreams. Through a partnership between the United States Department of Veterans Affairs and the USOC, the VA National Veterans Sports Programs & Special Events Office provides a monthly assistance allowance for disabled Veterans of the Armed Forces training in a Paralympic sport, as authorized by 38 U.S.C. § 322(d) and section 703 of the Veterans’ Benefits Improvement Act of 2008. Through the program the VA will pay a monthly allowance to a Veteran with a service-connected or non-service-connected disability if the Veteran meets the minimum VA Monthly Assistance Allowance (VMAA) Standard in his/her respective sport and sport class at a recognized competition. Athletes must have established training and competition plans and are responsible for turning in monthly and/or quarterly forms and reports in order to continue receiving the monthly assistance allowance. Additionally, an athlete must be U.S. citizen OR permanent resident to be eligible. Lastly, in order to be eligible for the VMAA athletes must undergo either national or international classification evaluation (and be found Paralympic sport eligible) within six months of being placed on the allowance pay list. -

2010 EG CVRS.Qxp
ELECTRICAL SOLUTIONS A. System Dome-Top ® Barb Ty Clamp Ties – Weather Resistant and Overview Heat Stabilized Nylon 6.6 B1. • Weather Resistant material has greater resistance to damage • Design allows for bundling before or after screwing clamp in place Cable Ties caused by ultraviolet light – indoor or outdoor use • Stainless steel locking barb provides consistent performance, • Heat stabilized material for high temperature applications reliability, and infinite adjustability through entire bundle range up to 239°F (115°C) – indoor use • Curved tip is easy to pick up from flat surfaces and allows faster B2. • Used to secure a cable bundle to another surface such as initial threading to speed installation Cable a control panel, communication rack, wall or ceiling Accessories B3. Stainless Steel Ties Straight Tip C1. Curved Tip Wiring Duct C2. Nominal Max. Min. Surface Raceway Hole Metric Bundle Loop Recommended Std. Std. Length Width Thickness Dia. Screw Screw Dia. Tensile Str. Installation Pkg. Ctn. Part Number In. mm In. mm In. mm In. mm Size Size In. mm Lbs. N Tool Qty. Qty. C3. Abrasion Weather Resistant Nylon 6.6 Protection Miniature Cross Section BC1M-S4-M0 4.6 117 .095 2.4 .046 1.2 .122 3.1 #4 M2.5 .90 23 18 80 GTS, GTSL, 1000 50000 C4. GS2B, PTS, BC2M-S4-M0 8.3 211 .095 2.4 .046 1.2 .122 3.1 #4 M2.5 2.00 51 18 80 1000 25000 Cable PPTS, STS2 Management Intermediate Cross Section BC1.5I-S8-M0 6.6 168 .141 3.6 .041 1.0 .174 4.4 #8 M4 1.50 38 40 178 GTS, GTSL, 1000 25000 GS2B, PTS, D1. -

Three to Six Pairs of Promising Sorghum Female Lines (A and B-Lines) Identified
Seventh framework programme Food, Agriculture and Fisheries, and Biotechnology Specific International Co-operation Actions Small or medium scale focused research project Grant Agreement n° 227422 WP1 Deliverable 1.7: Three to six pairs of promising sorghum female lines (A and B-lines) identified Composition of the consortium CIRAD ICRISAT EMBRAPA KWS IFEU UniBO UCSC ARC-GCI UANL WIP Promising new A- and B-lines selected out of the working collection In Table 1 you will find the most promising A-/B-lines with per se selection on line level. The females have reached now BC3-Level and could be tested on Hybrid-level in 2013. In Table 1 you will find the early vigor compared to the used Check-lines and also the value for the “General impression of the Line (GIMP)” before harvest. Table 2 gives the promising female lines identified by Cirad. Table 1: Best performing A-/B-Lines in comparison to the check rows (Check 1 -10) ID IDT INB_CODE GENOTYPE DER F_M PLASMA GENER EV 29_06 GIMP 1 ZM9-78291 Check 1 1Z1602A1 A1 S10 4 2,5 2 ZM0-30067 Check 2 1Z1064A1 A1 S10 3 2 3 ZM0-30077 Check 3 341x1Z1602B A1 F1 4 1 4 8N-8053 Check 4 341 A1 S10 4 2,5 5 8N-8070 Check 5 339 A1 S10 3 4 6 Z2Y-5504 Check 6 3Z10022A-BC211312 A1 S10 2,5 2 7 Z2Y-5502 Check 7 1Z2245A1 A1 S10 2 1,5 8 Z9N-9545 Check 8 352 A1 S10 2,5 2 9 Z2N-7020 Check 9 62 A1 S10 4 1 10 Z2N-7007 Check 10 64 A1 S10 4 1 11 ZC2-35012/001 E603-**121111 E603 P N S5 1,5 1 12 ZC2-35013/001 E603A1-**121111.1 1Z1064A1xE603 FD A1 BC3 1,5 53 ZC2-35092/001 E606-**281111 E606 P N S5 4 1,5 54 ZC2-35093/001 E606A1-**281111.1 -

Gut Microbiota Promotes Obesity-Associated Liver Cancer Through PGE 2-Mediated Suppression of Antitumor Immunity
Published OnlineFirst February 15, 2017; DOI: 10.1158/2159-8290.CD-16-0932 RESEARCH ARTICLE Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity Tze Mun Loo1, Fumitaka Kamachi1, Yoshihiro Watanabe1, Shin Yoshimoto2,3, Hiroaki Kanda4, Yuriko Arai1,2, Yaeko Nakajima-Takagi5, Atsushi Iwama5, Tomoaki Koga6, Yukihiko Sugimoto6, Takayuki Ozawa1, Masaru Nakamura1, Miho Kumagai1, Koichi Watashi7,8, Makoto M. Taketo9, Tomohiro Aoki10, Shuh Narumiya10,11,12, Masanobu Oshima12,13, Makoto Arita14,15,16,17, Eiji Hara2,12,18, and Naoko Ohtani1,16 ABSTRACT Obesity increases the risk of cancers, including hepatocellular carcinomas (HCC). However, the precise molecular mechanisms through which obesity promotes HCC development are still unclear. Recent studies have shown that gut microbiota may influence liver diseases by transferring its metabolites and components. Here, we show that the hepatic transloca- tion of obesity-induced lipoteichoic acid (LTA), a Gram-positive gut microbial component, promotes HCC development by creating a tumor-promoting microenvironment. LTA enhances the senescence- associated secretory phenotype (SASP) of hepatic stellate cells (HSC) collaboratively with an obesity- induced gut microbial metabolite, deoxycholic acid, to upregulate the expression of SASP factors and COX2 through Toll-like receptor 2. Interestingly, COX2-mediated prostaglandin E2 (PGE2) production suppresses the antitumor immunity through a PTGER4 receptor, thereby contributing to HCC progres- sion. -
Lcd & Dlp Projector Bulbs
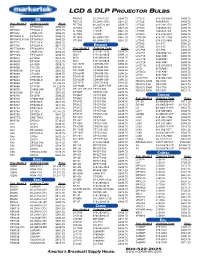
LCD & DLP PROJECTOR BULBS PB8263 5J.J2H01.001 $548.75 LV7320 610-289-6939 $498.75 3M PE5120 59.J9901.CG1 $561.25 LV7325 4638AA001 $498.75 Proj. Model # Ordering Code Price PE7765 60.J00720.001 $598.75 LV7345 610-293-2751 $498.75 540 EP7640ILK $486.25 SL703S 120VIP- $561.25 LV7350 7463A001AA $498.75 MP7630 EP7630LK $673.75 SL705S 120VIP- $561.25 LV7355 7463A001AA $498.75 MP7640 LAMP-029 $486.25 SL705X 120VIP- $561.25 LV7500 610-276-3010 $536.25 MP7640ILK EP7640ILK $486.25 VP150X 60.J0804.CB2 $561.25 LV7525E 610-282-2755 $498.75 MP7640ILK-S40 EP7640ILK $486.25 VT150X 60.J0804.CB2 $561.25 LV7545 610-293-5868 $536.25 MP7720 EP7720LK $836.25 LV7555 LV-LP17 $586.25 OXLIGHT MP7730 EP7630LK $673.75 B LV7565 LV-LP22 $623.75 Proj. Model # Ordering Code Price MP7730BAV EP7630BLK $723.75 LV-LP06 LV-LP06 $436.25 3600(A) 610-264-1943 $398.75 MP7740 LAMP-029 $486.25 LV-LP11 7463A001AA $498.75 3650 610-265-8828 $498.75 MP7740I EP7740ILK $498.75 LV-LP15 8441A001 $498.75 6000 610-265-8828 $498.75 MP8610 EP1830 $223.75 LV-LP18 9268A001 $498.75 6001 610-265-8828 $498.75 MP8620 EP1830 $223.75 LV-LP20 9431A001 $398.75 CD-450M CD450M-930 $586.25 MP8625 EP1890 $698.75 LV-S1-XL 610-293-8210 $498.75 CD-454 CP455M-930 $536.25 MP8630 CPL750 $461.25 LV-S2 8276A001 $498.75 CD-455M CP455M-930 $536.25 MP8635(B) EP-1625 $561.25 LV-S3(U) 9431A001 $398.75 CD-550M CD450M-930 $586.25 MP8640 CPL850 $498.75 LV-X2 8441A001 $498.75 CD-552M CP455M-930 $536.25 MP8647 EP8746LK $611.25 LV-X2THX 610-300-7267 $498.75 CD-555M CD450M-930 $586.25 MP8649 EP8749LK $511.25 REALIS SX50 RS-LP01 $623.75 CD-727X CD727X-930 $461.25 MP8650 EP1750 $1,223.75 REALIS SX6 RS-LP02 $623.75 CD-750M SP-LAMP-LP5F $561.25 MP8660 EP1750 $1,223.75 REALIS SX60 RS-LP03 $623.75 CP-13T, CP-33T CP13T-930 $498.75 MP8670 CP860-960 $523.75 REALIS X600 RS-LP02 $623.75 MP8725(B) EP-1625 $561.25 CP300T CP310T-930 $498.75 MP8740 EP2010 $586.25 CP305T CP13T-930 $498.75 CHRISTIE MP8745 CP860-960 $523.75 CP306T CP13T-930 $498.75 Proj. -

(VA) Veteran Monthly Assistance Allowance for Disabled Veterans
Revised May 23, 2019 U.S. Department of Veterans Affairs (VA) Veteran Monthly Assistance Allowance for Disabled Veterans Training in Paralympic and Olympic Sports Program (VMAA) In partnership with the United States Olympic Committee and other Olympic and Paralympic entities within the United States, VA supports eligible service and non-service-connected military Veterans in their efforts to represent the USA at the Paralympic Games, Olympic Games and other international sport competitions. The VA Office of National Veterans Sports Programs & Special Events provides a monthly assistance allowance for disabled Veterans training in Paralympic sports, as well as certain disabled Veterans selected for or competing with the national Olympic Team, as authorized by 38 U.S.C. 322(d) and Section 703 of the Veterans’ Benefits Improvement Act of 2008. Through the program, VA will pay a monthly allowance to a Veteran with either a service-connected or non-service-connected disability if the Veteran meets the minimum military standards or higher (i.e. Emerging Athlete or National Team) in his or her respective Paralympic sport at a recognized competition. In addition to making the VMAA standard, an athlete must also be nationally or internationally classified by his or her respective Paralympic sport federation as eligible for Paralympic competition. VA will also pay a monthly allowance to a Veteran with a service-connected disability rated 30 percent or greater by VA who is selected for a national Olympic Team for any month in which the Veteran is competing in any event sanctioned by the National Governing Bodies of the Olympic Sport in the United State, in accordance with P.L. -

BMD PLUG GAUGES Technical Guide Version 4 Content
BMD PLUG GAUGES Technical Guide Version 4 Content Page Page 3 DIATEST - Expertise for precision and safety 27 Indicator holders with adjustable spring pressure 4 Technical description 29 Electrical probe holders 9 Basic plug gauge types 30 Special gauge holders 10 Basic type - Standard 33 Holders for Analodig indicators 11 Basic type for through bores 34 Adapters 12 Basic type for blind bores 36 Right-angle attachments 13 Basic types with compressed air supply 37 Depth extensions 14 Plug gauges for automatic gauging 40 Depth stops 15 Special-purpose plug gauges 42 Small measurement fixtures 21 PA for measuring parallel wall gaps 46 Floating holders 22 Multiplane plug gauges 50 Spare parts 24 Indicator holders 51 General and technical abbreviations 2 Competence for precision and safety Safety through quality High-volume engineering does not work without precision. no matter whether it is a question of standard products To achieve highest possible safety in production, preci- or special solutions made to customer’s specifications. sion is necessary starting from design to final product. This is the company’s philosophy, carried out by an expe- Here the trademark DIATEST stands for quality. rienced staff: Highest quality at a fair cost effectiveness, Gauges with repeatabilities to 0.001 mm/0.00005“ combined with expert advice and absolute faithfulness and even better guarantee exact results. to deadlines in dealing with all DIATEST customers. For us this is a service taken for granted which our DIATEST bore gauges are manufactured according to DIATEST partners worldwide appreciate. DIN EN ISO 9001. Using state-of-the-art manufacturing engineering the highest quality standards are achieved. -

Layman's Guide to Classification
LAYMAN’S GUIDE TO CLASSIFICATION Swimming is the only sport that combines the conditions of limb loss, cerebral palsy (coordination and movement restrictions), spinal cord injury (weakness or paralysis involving any combination of the limbs) and other disabilities, such as Dwarfism; major joint restriction condition across classes. • Classes S1-S10 – are allocated to swimmers with a physical impairment • Classes S11-S13 – are allocated to swimmers with a visual impairment • Class S14 – is allocated to swimmers with an intellectual impairment • Class S15 – is allocated to swimmers with a hearing impairment • The Prefix S to the Class denotes the class for Freestyle, Backstroke and Butterfly • The Prefix SB to the class denotes the class for Breaststroke • The Prefix SM to the class denotes the class for Individual Medley The range is from the swimmers with a more severe impairment (S1, SB1, SM1) to those with the impairment (S10, SB9, SM10) In any one class some swimmers may start with a dive or in the water depending on their impairment. This is factored in when classifying an athlete. The following examples are only a guide - some conditions not mentioned here may also fit the following classes THE FUNCTIONAL CLASSIFICATION SYSTEM (FCS) PHYSICAL IMPAIRMENTS S1, SB1, SM1 Swimmers in this class would usually be wheelchair users and may have a higher dependency for their every day needs. Examples: Swimmers with very severe coordination problems in all four limbs or have no use of their legs, trunk, hands and minimal use of their shoulders only. Swimmers in this class usually only swim on their back.