Abstract Session I - MHC and Related Genes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Flow Reagents Single Color Antibodies CD Chart
CD CHART CD N° Alternative Name CD N° Alternative Name CD N° Alternative Name Beckman Coulter Clone Beckman Coulter Clone Beckman Coulter Clone T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells CD1a T6, R4, HTA1 Act p n n p n n S l CD99 MIC2 gene product, E2 p p p CD223 LAG-3 (Lymphocyte activation gene 3) Act n Act p n CD1b R1 Act p n n p n n S CD99R restricted CD99 p p CD224 GGT (γ-glutamyl transferase) p p p p p p CD1c R7, M241 Act S n n p n n S l CD100 SEMA4D (semaphorin 4D) p Low p p p n n CD225 Leu13, interferon induced transmembrane protein 1 (IFITM1). p p p p p CD1d R3 Act S n n Low n n S Intest CD101 V7, P126 Act n p n p n n p CD226 DNAM-1, PTA-1 Act n Act Act Act n p n CD1e R2 n n n n S CD102 ICAM-2 (intercellular adhesion molecule-2) p p n p Folli p CD227 MUC1, mucin 1, episialin, PUM, PEM, EMA, DF3, H23 Act p CD2 T11; Tp50; sheep red blood cell (SRBC) receptor; LFA-2 p S n p n n l CD103 HML-1 (human mucosal lymphocytes antigen 1), integrin aE chain S n n n n n n n l CD228 Melanotransferrin (MT), p97 p p CD3 T3, CD3 complex p n n n n n n n n n l CD104 integrin b4 chain; TSP-1180 n n n n n n n p p CD229 Ly9, T-lymphocyte surface antigen p p n p n -

Snipa Snpcard
SNiPAcard Block annotations Block info genomic range chr19:55,117,749-55,168,602 block size 50,854 bp variant count 74 variants Basic features Conservation/deleteriousness Linked genes μ = -0.557 [-4.065 – AC009892.10 , AC009892.5 , AC009892.9 , AC245036.1 , AC245036.2 , phyloP 2.368] gene(s) hit or close-by AC245036.3 , AC245036.4 , AC245036.5 , AC245036.6 , LILRA1 , LILRB1 , LILRB4 , MIR8061 , VN1R105P phastCons μ = 0.059 [0 – 0.633] eQTL gene(s) CTB-83J4.2 , LILRA1 , LILRA2 , LILRB2 , LILRB5 , LILRP1 μ = -0.651 [-4.69 – 2.07] AC008984.5 , AC008984.5 , AC008984.6 , AC008984.6 , AC008984.7 , AC008984.7 , AC009892.10 , AC009892.10 , AC009892.2 , AC009892.2 , AC009892.5 , AC010518.3 , AC010518.3 , AC011515.2 , AC011515.2 , AC012314.19 , AC012314.19 , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , -

An NK Cell-Activating Receptor with Inhibitory Potential Mathias Faure and Eric O
KIR2DL4 (CD158d), an NK Cell-Activating Receptor with Inhibitory Potential Mathias Faure and Eric O. Long This information is current as J Immunol 2002; 168:6208-6214; ; of September 25, 2021. doi: 10.4049/jimmunol.168.12.6208 http://www.jimmunol.org/content/168/12/6208 Downloaded from References This article cites 43 articles, 21 of which you can access for free at: http://www.jimmunol.org/content/168/12/6208.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on September 25, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2002 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology KIR2DL4 (CD158d), an NK Cell-Activating Receptor with Inhibitory Potential Mathias Faure and Eric O. Long1 KIR2DL4 (CD158d) is an unusual member of the killer cell Ig-like receptor family expressed in all NK cells and some T cells. KIR2DL4 activates the cytotoxicity of NK cells, despite the presence of an immunoreceptor tyrosine-based inhibition motif (ITIM) in its cytoplasmic tail. -

Natural Killer Cell Lymphoma Shares Strikingly Similar Molecular Features
Leukemia (2011) 25, 348–358 & 2011 Macmillan Publishers Limited All rights reserved 0887-6924/11 www.nature.com/leu ORIGINAL ARTICLE Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic cd T-cell lymphoma and is highly sensitive to a novel aurora kinase A inhibitor in vitro J Iqbal1, DD Weisenburger1, A Chowdhury2, MY Tsai2, G Srivastava3, TC Greiner1, C Kucuk1, K Deffenbacher1, J Vose4, L Smith5, WY Au3, S Nakamura6, M Seto6, J Delabie7, F Berger8, F Loong3, Y-H Ko9, I Sng10, X Liu11, TP Loughran11, J Armitage4 and WC Chan1, for the International Peripheral T-cell Lymphoma Project 1Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE, USA; 2Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE, USA; 3Departments of Pathology and Medicine, University of Hong Kong, Queen Mary Hospital, Hong Kong, China; 4Division of Hematology and Oncology, Department of Internal Medicine, University of Nebraska Medical Center, Omaha, NE, USA; 5College of Public Health, University of Nebraska Medical Center, Omaha, NE, USA; 6Departments of Pathology and Cancer Genetics, Aichi Cancer Center Research Institute, Nagoya University, Nagoya, Japan; 7Department of Pathology, University of Oslo, Norwegian Radium Hospital, Oslo, Norway; 8Department of Pathology, Centre Hospitalier Lyon-Sud, Lyon, France; 9Department of Pathology, Samsung Medical Center, Sungkyunkwan University, Seoul, Korea; 10Department of Pathology, Singapore General Hospital, Singapore and 11Penn State Hershey Cancer Institute, Pennsylvania State University College of Medicine, Hershey, PA, USA Natural killer (NK) cell lymphomas/leukemias are rare neo- Introduction plasms with an aggressive clinical behavior. -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

Human CD Marker Chart Reviewed by HLDA1 Bdbiosciences.Com/Cdmarkers
BD Biosciences Human CD Marker Chart Reviewed by HLDA1 bdbiosciences.com/cdmarkers 23-12399-01 CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD1a R4, T6, Leu6, HTA1 b-2-Microglobulin, CD74 + + + – + – – – CD93 C1QR1,C1qRP, MXRA4, C1qR(P), Dj737e23.1, GR11 – – – – – + + – – + – CD220 Insulin receptor (INSR), IR Insulin, IGF-2 + + + + + + + + + Insulin-like growth factor 1 receptor (IGF1R), IGF-1R, type I IGF receptor (IGF-IR), CD1b R1, T6m Leu6 b-2-Microglobulin + + + – + – – – CD94 KLRD1, Kp43 HLA class I, NKG2-A, p39 + – + – – – – – – CD221 Insulin-like growth factor 1 (IGF-I), IGF-II, Insulin JTK13 + + + + + + + + + CD1c M241, R7, T6, Leu6, BDCA1 b-2-Microglobulin + + + – + – – – CD178, FASLG, APO-1, FAS, TNFRSF6, CD95L, APT1LG1, APT1, FAS1, FASTM, CD95 CD178 (Fas ligand) + + + + + – – IGF-II, TGF-b latency-associated peptide (LAP), Proliferin, Prorenin, Plasminogen, ALPS1A, TNFSF6, FASL Cation-independent mannose-6-phosphate receptor (M6P-R, CIM6PR, CIMPR, CI- CD1d R3G1, R3 b-2-Microglobulin, MHC II CD222 Leukemia -

IL-2/15 Promoter Under Constitutive and KIR2DL4 Differential
Differential Transcription Factor Use by the KIR2DL4 Promoter under Constitutive and IL-2/15−Treated Conditions This information is current as Steven R. Presnell, Lei Zhang, Corrin N. Chlebowy, Ahmad of September 28, 2021. Al-Attar and Charles T. Lutz J Immunol 2012; 188:4394-4404; Prepublished online 30 March 2012; doi: 10.4049/jimmunol.1103352 http://www.jimmunol.org/content/188/9/4394 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2012/03/30/jimmunol.110335 Material 2.DC1 http://www.jimmunol.org/ References This article cites 62 articles, 39 of which you can access for free at: http://www.jimmunol.org/content/188/9/4394.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 28, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Differential Transcription Factor Use by the KIR2DL4 Promoter under Constitutive and IL-2/15–Treated Conditions Steven R. -
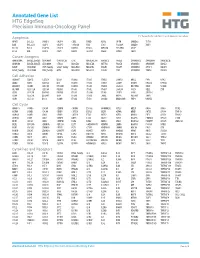
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel For Research Use Only. Not for use in diagnostic procedures. Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1 -

High-Resolution Analysis Identifies High Frequency of KIR-A
ORIGINAL RESEARCH published: 30 April 2021 doi: 10.3389/fimmu.2021.640334 High-Resolution Analysis Identifies High Frequency of KIR-A Haplotypes and Inhibitory Interactions of KIR With HLA Class I in Zhejiang Han Sudan Tao 1, Yanmin He 1, Katherine M. Kichula 2, Jielin Wang 1,JiHe1, Paul J. Norman 2* and Faming Zhu 1* 1 Blood Center of Zhejiang Province, Key Laboratory of Blood Safety Research of Zhejiang Province, Hangzhou, China, 2 Division of Biomedical Informatics and Personalized Medicine, and Department of Immunology and Microbiology, University Edited by: of Colorado Anschutz Medical Campus, Aurora, CO, United States Eleanor Riley, University of Edinburgh, United Kingdom Killer cell immunoglobulin-like receptors (KIR) interact with human leukocyte antigen (HLA) Reviewed by: class I molecules, modulating critical NK cell functions in the maintenance of human Kouyuki Hirayasu, health. Characterizing the distribution and characteristics of KIR and HLA allotype diversity Kanazawa University, Japan Roberto Biassoni, across defined human populations is thus essential for understanding the multiple Giannina Gaslini Institute (IRCCS), Italy associations with disease, and for directing therapies. In this study of 176 Zhejiang Han *Correspondence: individuals from Southeastern China, we describe diversity of the highly polymorphic KIR Faming Zhu [email protected] and HLA class I genes at high resolution. KIR-A haplotypes, which carry four inhibitory Paul J. Norman receptors specific for HLA-A, B or C, are known to associate with protection from infection [email protected] and some cancers. We show the Chinese Southern Han from Zhejiang are characterized Specialty section: by a high frequency of KIR-A haplotypes and a high frequency of C1 KIR ligands. -

Angiogenic Properties of NK Cells in Cancer and Other Angiogenesis-Dependent Diseases
cells Review Angiogenic Properties of NK Cells in Cancer and Other Angiogenesis-Dependent Diseases Dorota M. Radomska-Le´sniewska 1, Agata Białoszewska 1,* and Paweł Kami ´nski 2 1 Center for Biostructure Research, Department of Histology and Embryology, Medical University of Warsaw, 02-004 Warsaw, Poland; [email protected] 2 Department of Gynecology and Gynecological Oncology, Military Institute of Medicine, 04-349 Warsaw, Poland; [email protected] * Correspondence: [email protected] Abstract: The pathogenesis of many serious diseases, including cancer, is closely related to distur- bances in the angiogenesis process. Angiogenesis is essential for the progression of tumor growth and metastasis. The tumor microenvironment (TME) has immunosuppressive properties, which con- tribute to tumor expansion and angiogenesis. Similarly, the uterine microenvironment (UME) exerts a tolerogenic (immunosuppressive) and proangiogenic effect on its cells, promoting implantation and development of the embryo and placenta. In the TME and UME natural killer (NK) cells, which otherwise are capable of killing target cells autonomously, enter a state of reduced cytotoxicity or anergy. Both TME and UME are rich with factors (e.g., TGF-β, glycodelin, hypoxia), which support a conversion of NK cells to the low/non-cytotoxic, proangiogenic CD56brightCD16low phenotype. It is plausible that the phenomenon of acquiring proangiogenic and low cytotoxic features by NK cells is not only limited to cancer but is a common feature of different angiogenesis-dependent diseases (ADDs). In this review, we will discuss the role of NK cells in angiogenesis disturbances associated Citation: Radomska-Le´sniewska, D.M.; with cancer and other selected ADDs. Expanding the knowledge of the mechanisms responsible Białoszewska, A.; Kami´nski,P.