Insights Into Platypus Population Structure and History from Whole-Genome Sequencing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cape York Peninsula Marine Water Quality Synthesis
Cape York Peninsula Marine Water Quality Synthesis Technical Report for the CYP Water Quality Improvement Plan November 2015 0 Cover Photos: Coral assemblage (top left) and Crown of Thorns starfish (top right) at Little Unchartered Reef (Christina Howley). Bottom: Flood plume waters at Princess Charlotte Bay, approximately 14km offshore (north) from the Kennedy River mouth (Jeff Shellberg, 29/1/2013). Produced by Christina Howley (Howley Environmental Consulting) for South Cape York Catchments and Cape York NRM. This report was produced under contract to South Cape York Catchments (SCYC) with funding from the Australian Government for the Cape York Water Quality Improvement Plan. The scope of the report, as identified by SCYC and the WQIP Science Advisory Panel, was to synthesize the available water quality data from eastern CYP and to provide a comparison of the data from the northern, central and southern CYP regions and the GBR water quality guidelines. While every effort has been made to ensure that the contents of this publication are factually correct, the author does not accept responsibility for the accuracy or completeness of the contents. This publication is copyrighted and protected by the Australian Copyright Act 1968. All rights reserved. No part of this document may be used without the prior written permission of C. Howley and South Cape York Catchments. 1 EXECUTIVE SUMMARY The Cape York Peninsula (CYP) marine environment (far northern Great Barrier Reef) is considered to be in good condition compared to other parts of the Great Barrier Reef (GBR) or global coral reef ecosystems. This is attributed to a combination of factors, including good water quality due to lower levels of development within CYP catchments, as well as less pressure from recreational and commercial fishers, tourism and other boats. -

Orocline-Driven Transtensional Basins: Insights from the Lower Permian Manning Basin
PUBLICATIONS Tectonics RESEARCH ARTICLE Orocline-driven transtensional basins: Insights 10.1002/2015TC004021 from the Lower Permian Manning Key Points: Basin (eastern Australia) • Structures in the Manning Basin confirm the existence of the Llyam White1, Gideon Rosenbaum1, Charlotte M. Allen2, and Uri Shaanan1 Manning Orocline • The Manning Basin is an Early 1School of Earth Sciences, University of Queensland, Brisbane, Queensland, Australia, 2Institute for Future Environments, Permian sinistral transtensional pull-apart basin Queensland University of Technology, Brisbane, Queensland, Australia • Spatiotemporal relationships link basin formation to oroclinal bending Abstract The New England Orogen in eastern Australia exhibits an oroclinal structure, but its geometry and geodynamic evolution are controversial. Here we present new data from the southernmost part of the Supporting Information: • Supporting Information S1 oroclinal structure, the Manning Orocline, which supposedly developed in the Early Permian, contemporaneously and/or shortly after the deposition of the Lower Permian Manning Basin. New U-Pb detrital zircon data provide Correspondence to: a maximum depositional age of ~288 Ma. Structural evidence from rocks of the Manning Basin indicates that L. White, both bedding and preoroclinal fold axial planes are approximately oriented parallel to the trace of the Manning [email protected] Orocline. Brittle deformation was dominated by sinistral strike-slip faulting, particularly along a major fault zone (Peel-Manning Fault System), which is marked by the occurrence of a serpentinitic mélange, and separates Citation: tectonostratigraphic units of the New England Orogen. Our revised geological map shows that the Manning White, L., G. Rosenbaum, C. M. Allen, and U. Shaanan (2016), Orocline-driven Basin is bounded by faults and serpentinites, thus indicating that basin formation was intimately linked to transtensional basins: Insights from the deformation along the Peel-Manning Fault System. -

Government Gazette of the STATE of NEW SOUTH WALES Number 112 Monday, 3 September 2007 Published Under Authority by Government Advertising
6835 Government Gazette OF THE STATE OF NEW SOUTH WALES Number 112 Monday, 3 September 2007 Published under authority by Government Advertising SPECIAL SUPPLEMENT EXOTIC DISEASES OF ANIMALS ACT 1991 ORDER - Section 15 Declaration of Restricted Areas – Hunter Valley and Tamworth I, IAN JAMES ROTH, Deputy Chief Veterinary Offi cer, with the powers the Minister has delegated to me under section 67 of the Exotic Diseases of Animals Act 1991 (“the Act”) and pursuant to section 15 of the Act: 1. revoke each of the orders declared under section 15 of the Act that are listed in Schedule 1 below (“the Orders”); 2. declare the area specifi ed in Schedule 2 to be a restricted area; and 3. declare that the classes of animals, animal products, fodder, fi ttings or vehicles to which this order applies are those described in Schedule 3. SCHEDULE 1 Title of Order Date of Order Declaration of Restricted Area – Moonbi 27 August 2007 Declaration of Restricted Area – Woonooka Road Moonbi 29 August 2007 Declaration of Restricted Area – Anambah 29 August 2007 Declaration of Restricted Area – Muswellbrook 29 August 2007 Declaration of Restricted Area – Aberdeen 29 August 2007 Declaration of Restricted Area – East Maitland 29 August 2007 Declaration of Restricted Area – Timbumburi 29 August 2007 Declaration of Restricted Area – McCullys Gap 30 August 2007 Declaration of Restricted Area – Bunnan 31 August 2007 Declaration of Restricted Area - Gloucester 31 August 2007 Declaration of Restricted Area – Eagleton 29 August 2007 SCHEDULE 2 The area shown in the map below and within the local government areas administered by the following councils: Cessnock City Council Dungog Shire Council Gloucester Shire Council Great Lakes Council Liverpool Plains Shire Council 6836 SPECIAL SUPPLEMENT 3 September 2007 Maitland City Council Muswellbrook Shire Council Newcastle City Council Port Stephens Council Singleton Shire Council Tamworth City Council Upper Hunter Shire Council NEW SOUTH WALES GOVERNMENT GAZETTE No. -

IMPACTS of the UNPRECEDENTED 2019-20 BUSHFIRES on AUSTRALIAN ANIMALS NOVEMBER 2020 Acknowledgements
AUSTRALIA IMPACTS OF THE UNPRECEDENTED 2019-20 BUSHFIRES ON AUSTRALIAN ANIMALS NOVEMBER 2020 Acknowledgements WWF-Australia acknowledges the Traditional Owners of the land on which we work and their continuing connection to their lands, waters, and culture. We pay our respects to Elders – past and present, and their emerging leaders. WWF-Australia is part of the world’s largest conservation network. WWF-Australia has been working to create a world where people live in harmony with nature since 1978. WWF’s mission is to stop the degradation of the Earth’s CONTENTS natural environment and to build a future in which humans live in harmony with nature, by conserving the world’s biological diversity, ensuring that the use of renewable natural resources is sustainable, and promoting the EXECUTIVE SUMMARY 6 reduction of pollution and wasteful consumption. Prepared by Lily M van Eeden, Dale Nimmo, Michael BACKGROUND 10 Mahony, Kerryn Herman, Glenn Ehmke, Joris Driessen, James O’Connor, Gilad Bino, Martin Taylor and Chris 1.1 Fire in Australia 10 Dickman for WWF-Australia 1.2 The 2019-20 bushfire season 10 We are grateful to the researchers who provided data or feedback on the report. These include: 1.3 Scope of this study 12 • Eddy Cannella 1.3.1 Taxa included 14 • David Chapple 1.3.2 Study area 14 • Hugh Davies • Deanna Duffy 1.4 Limitations 17 • Hugh Ford • Chris Johnson 1. MAMMALS 18 • Brad Law 2.1 Methods 18 • Sarah Legge • David Lindenmayer 2.1.1 Most mammals 18 • Simon McDonald 2.1.2 Koalas 19 • Damian Michael 2.2 Results 22 • Harry Moore • Stewart Nichol 2.3 Caveats 22 • Alyson Stobo-Wilson • Reid Tingley 2. -

Spatial Patterns of Ne Sediment Supply and Transfer in the River Esk, North
Durham E-Theses Spatial patterns of ne sediment supply and transfer in the River Esk, North York Moors. Robinson, Katherine S. How to cite: Robinson, Katherine S. (2006) Spatial patterns of ne sediment supply and transfer in the River Esk, North York Moors., Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/2784/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk 2 Spatial patterns of fine sediment supply and transfer in the River Esk, North York Moors. The copyright of this thesis rests with the author or the university to which it was submitted. No quotation from it, or information derived from it may be published without the prior written consent of the author or university, and any information derived from it should be acknowledged. MSc (by Research) Katherine. S. Robinson Department of Geography Durham University September 2006 11 JUN 2007 Declaration This thesis is the result of my own work. -

Cape York Claims and Determinations
142°E 143°E 144°E 145°E Keirri Island Maururra Island ROUND ISLAND THURSDAY ISLAND CONSERVATION PARK ! ! Kaurareg MURALUG Aboriginal Muri Aboriginal Kaiwalagal AC Horn Land Trust Cape Land Trust Hammond Island York Mori CAPE YORK CLAIMS AND DETERMINATIONS Island POSSESSION Island CAPE YORK PENINSULA LAND TENURE EDITION 35 ISLAND Prepared by the Department of Natural Resources and Mines Townsville, Queensland, 6 June 2017 NATIONAL Ulrica Point PARK " Major Road Cape Cornwall Chandogoo Point Legend Homesteads/Roadhouse Minor Road Cliffy Point !( Population Centres SEISIA! River Boundary of CYP Region as referred !NEW MAPOON to in the CYP Heritage Act 2007 UMAGICO! ! Reef ! BAMAGA INJINOO Turtle Head Island DUNBAR Pastoral Holding Name Nature Refuge & Conservation Areas Slade Point Sharp Point Cape York Claims Cape York Determinations y Ck ck Sadd Point 11°S Ja 11°S y Classes of Land Tenure Apudthama k c Furze Point a Land Trust J JARDINE LandN ATIONALreserved- PARK Under ConservationNathe ture FREEHOLDINGincludingLEASE PURCHASELEASE SPECIAL RIVER Naaas tionaNaAct Park, Conserva l tionaor Park(Scientific) l tion theirforTena pay pricepurchaFREEHOLD these to - elects nt Jardine River RESOURCES Park. leawhichfreeho se, toconverts ldoncom pletionofpayments. RESERVE DENHAM GROUP Ussher NATIONAL PARK creaover tedAbo - N rigina ATIONAL(CYPAL) PARKland. l Land admLANDS- LEASE inisteredexcludingunderLand the Act JARDINE RIVER Point Traditionaformaareowners l (represented llybylanda trust) MiningHom esteaTenem d Lea ent ses. Vrilya Point NATIONAL PARK recognisedownersas ofland,thearea the being ma na gedaas NunderConservaNathe ain perpetuity tiona ture (CYPAL) Park tion l PERPETUincludingLEASES AL GRAZINGHOMESTEAD Act. PERPETUNON-COMPETITIVE LEASE, AL LEASE, Orford Ness N ON-COMPETITIVECONVLEASE Ongo ERTED - inglea seho ld CONSERVATIONRESOU PARK, Land RCESRESERVE Reserved- oragricultural e.g. -

Environmental Water Requirements for the Rubicon River
Environmental Water Requirements for The Rubicon River Tom Krasnicki Aquatic Ecologist Water Assessment and Planning Branch Water Resources Division DPIWE. Report Series WRA 02/01 May, 2002. Table of Contents ACKNOWLEDGEMENTS i GLOSSARY OF TERMS ii EXECUTIVE SUMMARY 1 1. INTRODUCTION 3 2. THE RUBICON RIVER 3 2.1 General Description 4 2.1.1 Catchment and Drainage System 3 2.1.2 Geomorphology and Geology 6 2.1.3 Climate and Rainfall 7 2.1.4 Vegetation 8 2.1.5 Land Use and Degradation 9 2.1.6 Port Sorell Estuary 9 2.1.7 Hydrology 11 2.2. Site Selection 13 2.2.1 The Rubicon River at Smith and Others Rd. 13 3. VALUES 15 3.1 Community Values 15 3.2 State Technical Values 17 3.3 Endangered species 18 3.4 Values Assessed 19 4. METHODOLOGY 20 4.1 Physical Habitat Data 20 4.2 Biological Data 21 4.2.1 Invertebrates 21 4.2.2 Fish 21 4.3 Hydraulic Simulation 21 4.4 Risk Analysis 22 5. RESULTS 24 5.1 Physical Habitat Data 24 5.2 Biological Data 25 5.3 Risk Analysis 26 6. DISCUSSION 29 6.1 Vertebrate Fauna 30 6.1.1 Mordacia mordax and Geotria australis 30 6.1.2 Gadopsis marmoratus 30 6.1.3 Pseudaphritis urvillii 31 6.1.4 Galaxias truttaceus and Galaxias maculatus 31 6.1.5 Galaxias brevipinnis and Neochanna cleaveri 31 6.1.6 Prototroctes maraena 32 6.1.7 Lovettia sealii and Retropinna tasmanica 32 6.1.8 Anguilla australis 32 6.1.9 Salmo trutta 32 6.1.10 Nannoperca australis and Perca fluviatilis 33 6.2 Invertebrate Fauna 33 6.2.1 Astacopsis gouldi 33 6.3 Flow Recommendations 34 6.3.1 Rubicon River at Smith and Others Rd. -

HERITAGE STOCK HORSE ASSESSMENTS (A-C) Updated: 1St November 2019 Approved Modern Breeds Unknown Thoroughbred Breeds After 1945 After 1960
HERITAGE STOCK HORSE ASSESSMENTS (A-C) Updated: 1st November 2019 Approved Modern Breeds Unknown Thoroughbred Breeds after 1945 after 1960 Name of Horse RegNo TB AB MB UB HSH Status Maximum Max 6.25% Max. 12.5% 100% Not Eligible 25% Combined Max 12.5% A BIT RASH 77780 Y 0 0 0 TB A COOL ACRE 234949 N 0 73.44 0 MB A GENTLE NUDGE BP-78750 Y 0 0 0 TB A LITTLE GOLD 77748 Y 0 0 0 TB A REWARD 74617 N 0 0 0 HSH A RIOT IN PARIS 74193 Y 0 0 0 TB A TOUCH OF CLASS 135153 Y 0 0 0 TB A TOUCH OF MAGIC 73306 N 0 0 0 HSH AACC CLEOPATRA 186032 N 0 43.75 0 LH AACC ELWOOD C2-188289 N 0 46.88 0 LH AACC EXPRESS C2-187868 N 0 46.88 0 LH AACC EXPRESSION 188286 N 0 12.5 0 LH AACC FEDERER 236049 N 0 25 12.5 LH AACC FORECAST C2-191980 N 0 53.13 0 MB AACC JEMIMA 197137 N 0 0 12.5 HSH AACC JODIE C2-178715 N 0 0 25 LH AACC RIPCURL C2-187870 N 0 59.38 0 MB AACC SPRITE 197922 N 0 50 0 MB AACC TRAVESTY 198605 N 0 40.63 6.25 LH AACC VALOAK 198022 N 0 34.38 12.5 LH AB RAY 35943 N 0 0 0 HSH ABA RAY 47264 N 0 0 0 HSH ABACIOMI 77441 Y 0 0 0 TB ABALMORES EXCEPTIONAL BELLA C1-199588 N 0 23.44 0 LH ABBA DABBA 30090 N 0 0 0 HSH ABBAGAIL 117120 N 0 0 0 HSH ABBALEE 33598 N 0 0 0 HSH ABBAMANDA 70990 N 0 0 0 HSH ABBCARS LITTLE WILDY 161304 N 4.69 0 0 HSH ABBELENA C1-237821 N 0 48.44 0 LH ABBELYSTIC 135950 N 0 0 0 HSH ABBER RAY DEE 103846 N 0 0 0 HSH ABBESS 130020 N 0 0 0 HSH ABBEY 448 N 0 0 0 HSH ABBEY BOY 56588 N 0 0 0 HSH ABBEY DOLL 106005 N 0 0 0 HSH ABBEY OKER 441 N 0 0 0 HSH ABBEY RUSH 152445 N 0 0 0 HSH ABBEY SKYE SM-161379 N 0 0 0 HSH ABBEY STAR 20213 N 0 0 0 HSH ABBEY SUE 63232 N 0 0 0 HSH ABBEY TANGO 20928 N 0 0 0 HSH Modern Breed - MBP 100%, MB 50-100% Heritage Stock Horse - HSH Approved Breed - ABP 100%, AB 50-100% Limited Heritage - LH (not eligible under other categories) Thoroughbred - TB Unknown Breed - UBP 100%, UB 50-100% Page 1 of 170 HERITAGE STOCK HORSE ASSESSMENTS (A-C) Updated: 1st November 2019 Approved Modern Breeds Unknown Thoroughbred Breeds after 1945 after 1960 Name of Horse RegNo TB AB MB UB HSH Status Maximum Max 6.25% Max. -
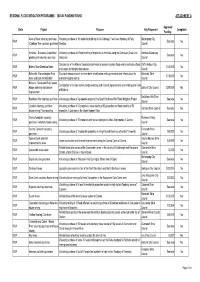
Answers to Questions on Notice
REGIONAL FLOOD MITIGATION PROGRAMME - 2003-04 FUNDING ROUND ATTACHMENT A Approved State Project Purpose Key Proponent Completed Funding Anama Street voluntary purchase Voluntary purchase of 16 residential buildings in the Cabbage Tree Creek floodway of Fairy Wollongong City NSW See note No (Cabbage Tree voluntary purchase) Meadow. Council Armidale - Dumaresq Creek flood Voluntary purchase and flood proofing of properties in Armidale along the Dumaresq Creek and Armidale Dumaresq NSW See note No proofing and voluntary purchase tributaries. Council Construciton of the Bakers Road detention basin to temporarily store flood waters and reduce flood Coffs Harbour City NSW Bakers Road Detention Basin $140,000 No discharges and heights downstream. Council Balranald - Murrumbidgee River Structural measures such as levee bank rehabilitation and augmentation and infrastructure for Balranald Shire NSW $110,000 No levee and bank rehabilitation overland flooding control. Council Bathurst - Macquarie River levees, Construction of multiple levees, bridge widening and channel improvements on the Macquarie River NSW bridge widening and channel Bathurst City Council $300,000 No at Bathurst. improvement Baulkham Hills Shire NSW Baulkham Hills voluntary purchase Voluntary purchase of 6 properties as part of the South Baulkham Hills Flood Mitigation Project. See note No Council Camden voluntary purchase / Voluntary purchase of 43 properties, house raising of 62 properties and flood proofing of 56 NSW Camden Shire Council See note No house raising / flood proofing properties in Camden on the Upper Nepean River. Casino floodplain voluntary Richmond Valley NSW Voluntary purchase of 12 properties and house raising of a further 26 properties in Casino. See note No purchase / voluntary house raising Council Central Tamworth voluntary Tamworth Shire NSW Voluntary purchase of 2 residential properties in a high hazard flood way of central Tamworth. -

December 2017
DECEMBER 2017 Newsletter of the Australian National Placenames Survey an initiative of the Australian Academy of Humanities, supported by the Geographical Names Board of NSW In search of the Basaltic Rock In August 1840 the Police District of Macquarie was Of the placenames listed in the original description all formally gazetted. It was described as: but one have endured. The exception is the Basaltic Rock, first described in an article on the opening of the Embracing the whole of the county of Macquarie, above north-eastern districts published in 1842: Crescent Head; bounded on the north by a line bearing west to a range heading Maria’s and Wilson’s Rivers, and before we quit New England, we must notice the by this range and a line from thence bearing south-westerly remarkable object described on the maps as the Basaltic to Werrekembe, at the source of the Hasting’s River, and by Rock. This is an insulated, and perpendicular column of the range heading the Apsley River to the Basaltic Rock, basalt, composed of hexagonal prisms closely cemented, and thence by a range to the range heading the River Peel; it forms an interesting feature of research to the geologist, on the south by the range heading Hunter’s River to the and a picturesque and singular object of contemplation to source of the Barnard, and by this river and the Manning the amateur tourist.5 to Farquhar’s Inlet on the sea shore, which forms the eastern boundary of the district to the point northward of Crescent Head aforesaid.1 Despite the reference to the ‘county of Macquarie’ no such county had yet been proclaimed so the creation of the police district was, in fact, the first time that the boundaries of the Macquarie district had been officially published. -

Gazetteer of Streams of Texas
DEPARTMENT OF THE INTERIOR FRANKLIN K. LANE, Secretary UNITED STATES GEOLOGICAL SURVEY GEORGE OTIS SMITH, Director Water-Supply Paper 448 GAZETTEER OF STREAMS OF TEXAS PREPARED UNDER THE DIRECTION OF GLENN A. GRAY WASHINGTON GOVERNMENT FEINTING OFFICE 1919 GAZETTEER OF STREAMS OF TEXAS. Prepared under the direction of GLENN A. GRAY. INTRODUCTION. The following pages contain a gazetteer of streams, lakes, and ponds as shown by the topographic maps of Texas which were pre pared by the United States Geological Survey and, in areas not covered by the topographic maps, by State of Texas county maps and the post-route map of Texas. For many streams a contour map of Texas, prepared in 1899 by Robert T. Hill, was consulted, as well as maps compiled by private surveys, engineering corporations, the State Board of Water Engineers, and the International Boundary Commission. An effort has been made to eliminate errors where practicable by personal reconnaissance. All the descriptions are based on the best available maps, and their accuracy therefore depends on that of the maps. Descriptions of streams in the central part of the State, adjacent to the Bio Grande above Brewster County, and in parts of Brewster, Terrell, Bowie, Casg, Btirleson, Brazos, Grimes, Washington, Harris, Bexar, Wichita, Wilbarger, Montague, Coke, and Graysoh counties were compiled by means of topographic maps and are of a good degree of accuracy. It should be understood, however, that all statements of elevation, length, and fall are roughly approximate. The Geological Survey topographic maps used are cited in the de scriptions of the streams and are listed below. -

The Riverfront Plan Asheville, North Carolina
The Riverfront Plan Asheville, North Carolina ^ ’■■i ^ S r " ^ ^W7st»-v5t : j r v : . roAjvncNu-J.'’ ®® Cover illustrations complivnents of Sandra Smathers-Jarman. Below, a view of Riverside Park and the French Broad River, ca. 1912.; photo courtesy of North Carolina Collection, Pack Memorial Public Library. Opposite, the same view in 1989; photo: J. Weiland. The Riverfront Plan Asheville, North Carolina April 26 - 29, 1989 A joint venture of: Urban Design Assistance Team, N.C. Chapter, The American Institute of Architects Community Assistance Team, N.C. Chapter, The American Society of Landscape Architects Sponsor: French Broad Riverfront Planning Cor Editor/Book Desigrt r.f * PREFACE For the greater part of the history of this Earth, atmospheric and AshevUle has reached a point In its history where the function and tectonic forces have forged the Blue Ridge Mountains and the character of the French Broad River must begin to respond to the Great Smokies. Over a much shorter period of time, plant and needs of its residents in a cultural and recreational way as well as animal ecosystems have flourished, creating the natural environ an economic way. It is a resource of such spectacular grandeur ment that surrounds Asheville today. Finally, at the end of a that the city can only benefit from its careful redevelopment. sequence of evolutionary events, man occupied the valleys and floodplains, seeking sustenance and shelter. The Riverfront Plan is a creation of many mmds, of citizens and professionals from the fields of architecture, landscape architec The French Broad River emerged from its source a long time ago in ture, and city and regional planning.