Identification of Key Mirna‑Mrna Pairs in Septic Mice by Bioinformatics Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CD81 Antibody (C-Term) Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP6631B
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 CD81 Antibody (C-term) Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP6631B Specification CD81 Antibody (C-term) - Product Information Application WB, IHC-P, FC,E Primary Accession P60033 Reactivity Human, Mouse Host Rabbit Clonality Polyclonal Isotype Rabbit Ig Antigen Region 176-203 CD81 Antibody (C-term) - Additional Information Gene ID 975 Other Names CD81 antigen, 26 kDa cell surface protein TAPA-1, Target of the antiproliferative antibody 1, Tetraspanin-28, Tspan-28, Western blot analysis of CD81 Antibody CD81, CD81, TAPA1, TSPAN28 (C-term) (Cat. #AP6631b) in mouse kidney(lane 1) and cerebellum(lane 2) tissue Target/Specificity lysates (35ug/lane). CD81 (arrow) was This CD81 antibody is generated from detected using the purified Pab. rabbits immunized with a KLH conjugated synthetic peptide between 176-203 amino acids from the C-terminal region of human CD81. Dilution WB~~1:1000 IHC-P~~1:10~50 FC~~1:10~50 Format Purified polyclonal antibody supplied in PBS with 0.09% (W/V) sodium azide. This antibody is prepared by Saturated Ammonium Sulfate (SAS) precipitation followed by dialysis against PBS. Storage Maintain refrigerated at 2-8°C for up to 2 Formalin-fixed and paraffin-embedded weeks. For long term storage store at -20°C in small aliquots to prevent freeze-thaw human brain tissue with CD81 Antibody cycles. (C-term), which was peroxidase-conjugated to the secondary antibody, followed by DAB Precautions staining. This data demonstrates the use of CD81 Antibody (C-term) is for research use this antibody for immunohistochemistry; Page 1/4 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 only and not for use in diagnostic or clinical relevance has not been evaluated. -

CD226 T Cells Expressing the Receptors TIGIT and Divergent Phenotypes of Human Regulatory
The Journal of Immunology Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226 Christopher A. Fuhrman,*,1 Wen-I Yeh,*,1 Howard R. Seay,* Priya Saikumar Lakshmi,* Gaurav Chopra,† Lin Zhang,* Daniel J. Perry,* Stephanie A. McClymont,† Mahesh Yadav,† Maria-Cecilia Lopez,‡ Henry V. Baker,‡ Ying Zhang,x Yizheng Li,{ Maryann Whitley,{ David von Schack,x Mark A. Atkinson,* Jeffrey A. Bluestone,‡ and Todd M. Brusko* Regulatory T cells (Tregs) play a central role in counteracting inflammation and autoimmunity. A more complete understanding of cellular heterogeneity and the potential for lineage plasticity in human Treg subsets may identify markers of disease pathogenesis and facilitate the development of optimized cellular therapeutics. To better elucidate human Treg subsets, we conducted direct transcriptional profiling of CD4+FOXP3+Helios+ thymic-derived Tregs and CD4+FOXP3+Helios2 T cells, followed by comparison with CD4+FOXP32Helios2 T conventional cells. These analyses revealed that the coinhibitory receptor T cell Ig and ITIM domain (TIGIT) was highly expressed on thymic-derived Tregs. TIGIT and the costimulatory factor CD226 bind the common ligand CD155. Thus, we analyzed the cellular distribution and suppressive activity of isolated subsets of CD4+CD25+CD127lo/2 T cells expressing CD226 and/or TIGIT. We observed TIGIT is highly expressed and upregulated on Tregs after activation and in vitro expansion, and is associated with lineage stability and suppressive capacity. Conversely, the CD226+TIGIT2 population was associated with reduced Treg purity and suppressive capacity after expansion, along with a marked increase in IL-10 and effector cytokine production. These studies provide additional markers to delineate functionally distinct Treg subsets that may help direct cellular therapies and provide important phenotypic markers for assessing the role of Tregs in health and disease. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

CD Markers Are Routinely Used for the Immunophenotyping of Cells
ptglab.com 1 CD MARKER ANTIBODIES www.ptglab.com Introduction The cluster of differentiation (abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules. So-called CD markers are routinely used for the immunophenotyping of cells. Despite this use, they are not limited to roles in the immune system and perform a variety of roles in cell differentiation, adhesion, migration, blood clotting, gamete fertilization, amino acid transport and apoptosis, among many others. As such, Proteintech’s mini catalog featuring its antibodies targeting CD markers is applicable to a wide range of research disciplines. PRODUCT FOCUS PECAM1 Platelet endothelial cell adhesion of blood vessels – making up a large portion molecule-1 (PECAM1), also known as cluster of its intracellular junctions. PECAM-1 is also CD Number of differentiation 31 (CD31), is a member of present on the surface of hematopoietic the immunoglobulin gene superfamily of cell cells and immune cells including platelets, CD31 adhesion molecules. It is highly expressed monocytes, neutrophils, natural killer cells, on the surface of the endothelium – the thin megakaryocytes and some types of T-cell. Catalog Number layer of endothelial cells lining the interior 11256-1-AP Type Rabbit Polyclonal Applications ELISA, FC, IF, IHC, IP, WB 16 Publications Immunohistochemical of paraffin-embedded Figure 1: Immunofluorescence staining human hepatocirrhosis using PECAM1, CD31 of PECAM1 (11256-1-AP), Alexa 488 goat antibody (11265-1-AP) at a dilution of 1:50 anti-rabbit (green), and smooth muscle KD/KO Validated (40x objective). alpha-actin (red), courtesy of Nicola Smart. PECAM1: Customer Testimonial Nicola Smart, a cardiovascular researcher “As you can see [the immunostaining] is and a group leader at the University of extremely clean and specific [and] displays Oxford, has said of the PECAM1 antibody strong intercellular junction expression, (11265-1-AP) that it “worked beautifully as expected for a cell adhesion molecule.” on every occasion I’ve tried it.” Proteintech thanks Dr. -

The Discovery and Regulation of Modes of Exocytosis Through the Lens of Computer Vision
THE DISCOVERY AND REGULATION OF MODES OF EXOCYTOSIS THROUGH THE LENS OF COMPUTER VISION Fabio Urbina A thesis submitted to the faculty at the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the department of Cell Biology and Physiology in the School of Medicine. Chapel Hill 2020 Approved by: Stephanie Gupton Patrick Brennwald Doug Cyr Keith Burridge Shawn Gomez © 2020 Fabio Urbina ALL RIGHTS RESERVED ii ABSTRACT Fabio Urbina: Discovery and Regulation of the Modes of Exocytosis through the lens of computer vision (Under the direction of Stephanie Gupton) The formation of the nervous system involves establishing complex networks of synaptic connections between proper partners, which requires the rapid expansion of the plasma membrane surface area as neurons grow. Critical to the expansion of the plasma membrane is exocytic vesicle fusion, a regulated mechanism driven by soluble N-ethylmaleimide-sensitive factor attachment proteins receptors (SNAREs). Multiple modes of exocytosis have been proposed, with full-vesicle fusion (FVF) and kiss-and-run (KNR) being the best described. The basis of SNARE-mediated fusion, the opening of a fusion pore, and its contribution to plasma membrane expansion remains enigmatic, as vesicle fusion is spatially small and temporally fast. We exploited TIRF microscopy to image VAMP-pHluorin mediated exocytosis in murine embryonic cortical neurons and developed computer-vision software and statistical tools to perform unbiased, efficient identification of exocytic events and uncover spatiotemporal aspects of exocytosis during neuron development. We further developed novel classification algorithms to describe. and classify individual exocytic events. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

The VE-Cadherin/Amotl2 Mechanosensory Pathway Suppresses Aortic In�Ammation and the Formation of Abdominal Aortic Aneurysms
The VE-cadherin/AmotL2 mechanosensory pathway suppresses aortic inammation and the formation of abdominal aortic aneurysms Yuanyuan Zhang Karolinska Institute Evelyn Hutterer Karolinska Institute Sara Hultin Karolinska Institute Otto Bergman Karolinska Institute Maria Forteza Karolinska Institute Zorana Andonovic Karolinska Institute Daniel Ketelhuth Karolinska University Hospital, Stockholm, Sweden Joy Roy Karolinska Institute Per Eriksson Karolinska Institute Lars Holmgren ( [email protected] ) Karolinska Institute Article Keywords: arterial endothelial cells (ECs), vascular disease, abdominal aortic aneurysms Posted Date: June 15th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-600069/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License The VE-cadherin/AmotL2 mechanosensory pathway suppresses aortic inflammation and the formation of abdominal aortic aneurysms Yuanyuan Zhang1, Evelyn Hutterer1, Sara Hultin1, Otto Bergman2, Maria J. Forteza2, Zorana Andonovic1, Daniel F.J. Ketelhuth2,3, Joy Roy4, Per Eriksson2 and Lars Holmgren1*. 1Department of Oncology-Pathology, BioClinicum, Karolinska Institutet, Stockholm, Sweden. 2Department of Medicine Solna, BioClinicum, Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden. 3Department of Cardiovascular and Renal Research, Institutet of Molecular Medicine, Univ. of Southern Denmark, Odense, Denmark 4Department of Molecular Medicine and Surgery, Karolinska Institutet, Karolinska University Hospital, Stockholm, -

Time-Resolved Systems Analysis of Virus Infection Fate Regulation
Time-resolved systems analysis of virus infection fate regulation Mireia Pedragosa Marín TESI DOCTORAL UPF / 2018 DIRECTORS DE LA TESI Dr. Andreas Meyerhans i Dr. Jordi Argilaguet DEPARTAMENT DE CIÈNCIES EXPERIMENTALS I DE LA SALUT Als que estimo, i a les “Surullotes”, ACKNOWLEDGEMENTS Sembla mentida però per fi ha arribat el moment d’entregar i defensar la tesi. No m’ho puc ni creure… Així que toca agraïr a tots i totes els/les que han fet que això hagi estat posible. To start with, I want to thank my supervisor Dr. Andreas Meyerhans, not only for providing a lab and money for the experiments, but also for his help during the thesis. I know I am not always easy and that I have still lots of things to learn… but I enjoyed (and will continue enjoying) the process. Jordi, coneixent-me saps que, de tota la tesi, el que més m’està costant és aquest apartat… així que seré breu com sempre. INFINITES GRÀCIES PER TOT. Necessito una tesi sencera per poder agraïr-te tot el que has fet (I segur que hauràs de seguir fent, ho sento). Gràcies pel suport moral, mental i físic. I per cert, no m’oblido de la paella… To all my labmates from the past and from the present. Gracielita (gracias por enseñarme Paraguayo Y por hacer del lab una fiesta), Vale, Eva, Kat, Mie, Javier, and the people from Juana’s lab. You make from everyday a different and happy day, really. A l’Eva, l’Erika, l’Òscar i a l’Àlex per la seva ajuda en els projectes de flow, que no sempre surt tot com una vol i per les xerrades “no tècniques” que fan les hores de flow més curtes. -

NKTR-214 2017SITC Poster-P77
The Novel IL-2 Cytokine Immune Agonist NKTR-214 Harnesses the Adaptive and Innate Immune System for the Treatment of Solid Cancers Salah Eddine Bentebibel1, Chantale Bernatchez1, Cara Haymaker1, Michael Hurwitz3, Patrick Hwu1, Mario Sznol3, Nizar Tannir1, Anthony Conley1, Harriet Kluger3, Sandra Aung2, Michael Imperiale2, Mary Tagliaferri2, Christie Fanton2, Ernesto Iacucci2, Jonathan Zalevsky2, Ute Hoch2, Adi Diab1 1The University of Texas MD Anderson Cancer Center, Houston, TX, United States of America, 2Nektar Therapeutics, San Francisco, CA, United States of America, 3Yale School of Medicine, New Haven, CT, United States of America BACKGROUND RESULTS NKTR-214 Figure 1. NKTR-214 Mechanism of Action Figure 5. NKTR-214 induces activating markers and co-inhibitory Figure 10. Increase in T cell clonality in tumors after treatment • NKTR-214 is a CD122-biased cytokine agonist + NKTR-214 2-PEG 1-PEG receptors in proliferating CD4 T cells in peripheral blood with NKTR-214 conjugated with multiple releasable chains of (6-PEG) Active Cytokine Active Cytokine PD-1 CTLA-4 OX40 ICOS Irreversible Irreversible 15 40 20 80 5 polyethylene glycol (PEG) designed to provide Release Release • An increase in overall T cell n=11 n=15 n=9 n=15 4 (%CD4) sustained signaling through the heterodimeric + 30 60 3 (%CD4) 15 (%CD4) + (%CD4) + clonality observed in 8/12 + 10 + 2 IL-2 receptor pathway (IL-2R) to preferentially 20 1 PD-1 10 40 patients after NKTR-214 OX40 ICOS + + 10 + CTLA-4 LEGEND: + + 0 activate and expand effector CD8 T and NK 5 α Ki-67 treatment 1 NKTR-214 – Inactive 5 20 -1 Melanoma Ki67 Ki-67 5 Ki-67 + Ki-67 cells over Tregs (Figure 1). -

BD Pharmingen™ Purified Mouse Anti-CD247 (Py142)
BD Pharmingen™ Technical Data Sheet Purified Mouse anti-CD247 (pY142) Product Information Material Number: 558402 Alternate Name: CD3ζ Size: 0.1 mg Concentration: 0.5 mg/ml Clone: K25-407.69 Immunogen: Phosphorylated Human CD3ζ Peptide Isotype: Mouse IgG2a, κ Reactivity: QC Testing: Human Target MW: 21 kDa Storage Buffer: Aqueous buffered solution containing ≤0.09% sodium azide. Description The T cell receptor (TCR), expressed by thymocytes and T lymphocytes, is a multi-component cell-surface complex responsible for recognizing antigen in the context of MHC molecules. The antigen-specific binding component of the TCR, Ti, is a heterodimer of the variable Ig-like subunits a and b or g and d. Ti is non-covalently associated with an invariant set of molecules referred to as the CD3 polypeptides, g, d, e, and ζ. The CD3 z polypeptide (CD3z) was named CD247 at the 7th Human Leukocyte Differentiation Antigens Workshop. CD3 appears early in thymocyte differentiation and remains expressed on all mature T lymphocytes. After antigen recognition by the TCR, CD3z is the primary intracellular signal transducing subunit. It contains three ITAMs (Immunoreceptor Tyrosine-based Activation Motifs), each of which contains a pair of tyrosine residues that are phosphorylated by Lck and Fyn and are required for signal propagation. The molecular weight of CD3z is 16 kDa, and it is also observed as 32-kDa homodimers or as heterodimers with the g chain of Fc receptors. Upon phosphorylation, the CD3z monomer undergoes an apparent shift in electrophoretic mobility up to 21 kDa. The K25-407.69 monoclonal antibody recognizes the phosphorylated tyrosine 142 (pY142) in the third ITAM domain of human CD3z (CD247). -

CD81 Antibody Purified Mouse Monoclonal Antibody (Mab) Catalog # Am8557b
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 CD81 Antibody Purified Mouse Monoclonal Antibody (Mab) Catalog # AM8557b Specification CD81 Antibody - Product Information Application WB, IHC,E Primary Accession P60033 Other Accession P60034 Reactivity Human, Rat Host Mouse Clonality monoclonal Isotype IgG1,k CD81 Antibody - Additional Information Gene ID 975 Other Names CD81 antigen, 26 kDa cell surface protein TAPA-1, Target of the antiproliferative antibody 1, Tetraspanin-28, Tspan-28, Anti-CD81 Antibody at 1:4000 dilution + rat CD81, CD81, TAPA1, TSPAN28 cerebellum whole cell lysate Lysates/proteins at 20 µg per lane. Secondary Goat Target/Specificity Anti-Mouse IgG, (H+L), Peroxidase This CD81 antibody is generated from a conjugated at 1/10000 dilution. Predicted mouse immunized with recombinant protein band size : 26 kDa Blocking/Dilution buffer: from the human region of human CD81. 5% NFDM/TBST. Dilution WB~~1:2000 IHC~~1:1000 Format Purified monoclonal antibody supplied in PBS with 0.09% (W/V) sodium azide. This antibody is purified through a protein G column, followed by dialysis against PBS. Storage Maintain refrigerated at 2-8°C for up to 2 weeks. For long term storage store at -20°C in small aliquots to prevent freeze-thaw cycles. Precautions CD81 Antibody is for research use only and not for use in diagnostic or therapeutic Anti-CD81 Antibody at 1:4000 dilution + procedures. human testis whole cell lysate Lysates/proteins at 20 µg per lane. Secondary Goat Anti-Mouse IgG, (H+L), Page 1/4 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 CD81 Antibody - Protein Information Peroxidase conjugated at 1/10000 dilution. -
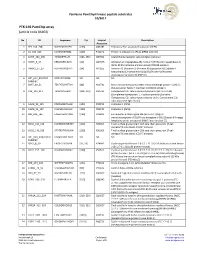
PTK-196 Pamchip Array (Article Code 86402)
PamGene PamChip4 kinase peptide substrates 10/2017 PTK-196 PamChip array (article code 86402) No ID Sequence Tyr Uniprot Description Accession 1 EFS_246_258 GGTDEGIYDVPLL [253] O43281 Embryonal Fyn-associated substrate (HEFS). 2 41_654_666 LDGENIYIRHSNL [660] P11171 Protein 4.1 (Band 4.1) (P4.1) (EPB4.1) (4.1R). 3 ACHD_383_395 YISKAEEYFLLKS [383, 390] Q07001 Acetylcholine receptor subunit delta precursor. 4 AMPE_5_17 EREGSKRYCIQTK [12] Q07075 Glutamyl aminopeptidase (EC 3.4.11.7) (EAP) (Aminopeptidase A) (APA) (Differentiation antigen gp160) (CD249 antigen). 5 ANXA2_17_29 HSTPPSAYGSVKA [24] P07355 Annexin A2 (Annexin-2) (Annexin II) (Lipocortin II) (Calpactin I heavychain) (Chromobindin-8) (p36) (Protein I) (Placental anticoagulantprotein IV) (PAP-IV). 6 ART_004_EAIYAAP EAIYAAPFAKKK NA NA NA FAKKKXC 7 B3AT_39_51 TEATATDYHTTSH [46] P02730 Band 3 anion transport protein (Anion exchange protein 1) (AE 1) (Solute carrier family 4 member 1) (CD233 antigen). 8 C1R_199_211 TEASGYISSLEYP [204, 210] P00736 Complement C1r subcomponent precursor (EC 3.4.21.41) (Complementcomponent 1, r subcomponent) [Contains: Complement C1r subcomponentheavy chain; Complement C1r subcomponent light chain]. 9 CALM_93_105 FDKDGNGYISAAE [100] P0DP23 Calmodulin (CaM). 10 CALM_95_107 KDGNGYISAAELR [100] P0DP23 Calmodulin (CaM). 11 CBL_693_705 EGEEDTEYMTPSS [700] P22681 E3 ubiquitin-protein ligase CBL (EC 6.3.2.-) (Signal transductionprotein CBL) (Proto-oncogene c-CBL) (Casitas B-lineage lymphoma proto-oncogene) (RING finger protein 55). 12 CD3Z_116_128 KDKMAEAYSEIGM [123] P20963 T-cell surface glycoprotein CD3 zeta chain precursor (T-cell receptorT3 zeta chain) (CD247 antigen). 13 CD3Z_146_158 STATKDTYDALHM [153] P20963 T-cell surface glycoprotein CD3 zeta chain precursor (T-cell receptorT3 zeta chain) (CD247 antigen). 14 ART_003_EAI(pY)AAP EAI(pY)AAPFAKKK NA NA NA FAKKKXC 15 CDK2_8_20 EKIGEGTYGVVYK [15, 19] P24941 Cyclin-dependent kinase 2 (EC:2.7.11.22) Cell division protein kinase 2 (EC 2.7.11.22) (p33 protein kinase).