Genotype-Phenotype Association Study Via New Multi-Task Learning Model
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

This Is a Sample Copy, Not to Be Reproduced Or Sold
Startup Business Chinese: An Introductory Course for Professionals Textbook By Jane C. M. Kuo Cheng & Tsui Company, 2006 8.5 x 11, 390 pp. Paperback ISBN: 0887274749 Price: TBA THIS IS A SAMPLE COPY, NOT TO BE REPRODUCED OR SOLD This sample includes: Table of Contents; Preface; Introduction; Chapters 2 and 7 Please see Table of Contents for a listing of this book’s complete content. Please note that these pages are, as given, still in draft form, and are not meant to exactly reflect the final product. PUBLICATION DATE: September 2006 Workbook and audio CDs will also be available for this series. Samples of the Workbook will be available in August 2006. To purchase a copy of this book, please visit www.cheng-tsui.com. To request an exam copy of this book, please write [email protected]. Contents Tables and Figures xi Preface xiii Acknowledgments xv Introduction to the Chinese Language xvi Introduction to Numbers in Chinese xl Useful Expressions xlii List of Abbreviations xliv Unit 1 问好 Wènhǎo Greetings 1 Unit 1.1 Exchanging Names 2 Unit 1.2 Exchanging Greetings 11 Unit 2 介绍 Jièshào Introductions 23 Unit 2.1 Meeting the Company Manager 24 Unit 2.2 Getting to Know the Company Staff 34 Unit 3 家庭 Jiātíng Family 49 Unit 3.1 Marital Status and Family 50 Unit 3.2 Family Members and Relatives 64 Unit 4 公司 Gōngsī The Company 71 Unit 4.1 Company Type 72 Unit 4.2 Company Size 79 Unit 5 询问 Xúnwèn Inquiries 89 Unit 5.1 Inquiring about Someone’s Whereabouts 90 Unit 5.2 Inquiring after Someone’s Profession 101 Startup Business Chinese vii Unit -

(Marco) Nie Ing, Northwestern University, Evanston, Illinois 60208, USA
Address: Department of Civil and Environmental Engineer- Yu (Marco) Nie ing, Northwestern University, Evanston, Illinois 60208, USA. Curriculum Vitae Phone: +1 847-467-0502 Email: [email protected] October 2018 WWW: http://www.mccormick.northwestern.edu/research- faculty/directory/profiles/nie-yu.html Appointments 2017 - present Professor Civil and Environmental Engineering, Northwestern University, affiliated with Northwestern University Transportation Center 2012 - 2017 Associate Professor Civil and Environmental Engineering, Northwestern University, affiliated with Northwestern University Transportation Center 2015 - present Adjunct Professor School of Transportation and Logistics, Southwestern Jiaotong University, Chengdu, China 2006 - 2012 Assistant Professor Civil and Environmental Engineering, Northwestern University, affiliated with Northwestern University Transportation Center Education and Qualifications 1999 B.Sc.(Hons) Tsinghua University Civil Engineering 2001 M.Eng. National University of Singapore Civil and Environmental Engineering 2006 Ph.D. University of California, Davis Civil and Environmental Engineering Honors and Awards 2018 Stella Dafermos Best Paper Award TRB Transportation Network Mod- eling Committee 2006 -2009 Louis Berger Junior Chair Northwestern University 2007-2008 Searle Junior Faculty Fellow Northwestern University 2003-2004 John Muir Fellowship University of California, Davis 1999 Outstanding Student Award Tsinghua University 1998 United Technology RongHong Scholarship Tsinghua University Publications By October 2018, I have authored or co-authored 78 articles in peer-reviewed journals, including 28 in Transportation Research Part B, 3 in Transportation Science. My H-Index is 23 according to Scopus 1 and 30 according to Google Scholar. Refereed articles 1. Chen, P. and Y. M. Nie (2018). Optimal Design of Demand Adaptive Paired-Line Hybrid Transit: Case of Radial Route Structure. Transportation Research Part E 110, 71–89. -

Effect of Trace Rare Earth Element Er on Al-Zn-Mg Alloy
Effect of trace rare earth element Er on Al-Zn-Mg alloy XU Guo-fu(徐国富)1, MOU Shen-zhou(牟申周)1, YANG Jun-jun(杨军军)2, JIN Tou-nan(金头男)3, NIE Zuo-ren(聂祚仁)3, YIN Zhi-min(尹志民)1 1. School of Materials Science and Engineering, Central South University, Changsha 410083, China; 2. Ltd AT&M, Central Iron & Steel Research Institute, Beijing 100039, China; 3. School of Materials Science and Engineering, Beijing University of Technology, Beijing 100022, China Received 24 January 2006; accepted 26 April 2006 Abstract: Al-6Zn-2Mg and Al-6Zn-2Mg-0.4Er alloys were prepared by cast metallurgy. The effects of trace Er on the mechanical properties, recrystallization behavior and age-hardening characteristic of Al-Zn-Mg alloy were studied. The effect of Er on microstructures was also studied by OM, XRD, SEM, EDS and TEM. The results show that the addition of Er on Al-6Zn-2Mg alloy is capable of refining grains obviously. The addition of Er can improve the strength considerably by strengthening mechanisms of precipitation and grain refinement. With the addition of Er into Al-6Zn-2Mg alloy, the aging process is quickened and the age-hardening effect is heightened. Er additive can retard the recrystallizing behavior of Al-6Zn-2Mg alloy and cause the increase of recrystallization temperature due to the pinning effect of fine dispersed Al3Er precipitates on dislocations and subgrain boundaries. Key words: erbium(Er); Al-Zn-Mg alloy; Al3Er; precipitation strengthening; fine grain strengthening; mechanical properties; recrystallization 1 Introduction 2 Experimental Remarkable effects of rear-earth elements on The alloys are prepared by cast metallurgy with aluminium alloys, such as eliminating impurities, high purity aluminium (99.99%), industrially pure zinc purifying melt, refining as-cast structure, retarding (99.9%), industrially pure magnesium (99.9%), and recrystallization and refining precipitated phases, have master alloy Al-6.2Er. -

Jingjiao Under the Lenses of Chinese Political Theology
religions Article Jingjiao under the Lenses of Chinese Political Theology Chin Ken-pa Department of Philosophy, Fu Jen Catholic University, New Taipei City 24205, Taiwan; [email protected] Received: 28 May 2019; Accepted: 16 September 2019; Published: 26 September 2019 Abstract: Conflict between religion and state politics is a persistent phenomenon in human history. Hence it is not surprising that the propagation of Christianity often faces the challenge of “political theology”. When the Church of the East monk Aluoben reached China in 635 during the reign of Emperor Tang Taizong, he received the favorable invitation of the emperor to translate Christian sacred texts for the collections of Tang Imperial Library. This marks the beginning of Jingjiao (oY) mission in China. In historiographical sense, China has always been a political domineering society where the role of religion is subservient and secondary. A school of scholarship in Jingjiao studies holds that the fall of Jingjiao in China is the obvious result of its over-involvement in local politics. The flaw of such an assumption is the overlooking of the fact that in the Tang context, it is impossible for any religious establishments to avoid getting in touch with the Tang government. In the light of this notion, this article attempts to approach this issue from the perspective of “political theology” and argues that instead of over-involvement, it is rather the clashing of “ideologies” between the Jingjiao establishment and the ever-changing Tang court’s policies towards foreigners and religious bodies that caused the downfall of Jingjiao Christianity in China. This article will posit its argument based on the analysis of the Chinese Jingjiao canonical texts, especially the Xian Stele, and takes this as a point of departure to observe the political dynamics between Jingjiao and Tang court. -
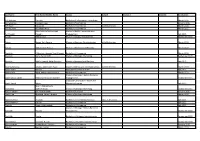
Last Name First Name/Middle Name Course Award Course 2 Award 2 Graduation
Last Name First Name/Middle Name Course Award Course 2 Award 2 Graduation A/L Krishnan Thiinash Bachelor of Information Technology March 2015 A/L Selvaraju Theeban Raju Bachelor of Commerce January 2015 A/P Balan Durgarani Bachelor of Commerce with Distinction March 2015 A/P Rajaram Koushalya Priya Bachelor of Commerce March 2015 Hiba Mohsin Mohammed Master of Health Leadership and Aal-Yaseen Hussein Management July 2015 Aamer Muhammad Master of Quality Management September 2015 Abbas Hanaa Safy Seyam Master of Business Administration with Distinction March 2015 Abbasi Muhammad Hamza Master of International Business March 2015 Abdallah AlMustafa Hussein Saad Elsayed Bachelor of Commerce March 2015 Abdallah Asma Samir Lutfi Master of Strategic Marketing September 2015 Abdallah Moh'd Jawdat Abdel Rahman Master of International Business July 2015 AbdelAaty Mosa Amany Abdelkader Saad Master of Media and Communications with Distinction March 2015 Abdel-Karim Mervat Graduate Diploma in TESOL July 2015 Abdelmalik Mark Maher Abdelmesseh Bachelor of Commerce March 2015 Master of Strategic Human Resource Abdelrahman Abdo Mohammed Talat Abdelziz Management September 2015 Graduate Certificate in Health and Abdel-Sayed Mario Physical Education July 2015 Sherif Ahmed Fathy AbdRabou Abdelmohsen Master of Strategic Marketing September 2015 Abdul Hakeem Siti Fatimah Binte Bachelor of Science January 2015 Abdul Haq Shaddad Yousef Ibrahim Master of Strategic Marketing March 2015 Abdul Rahman Al Jabier Bachelor of Engineering Honours Class II, Division 1 -

Video Recovery Via Learning Variation and Consistency of Images
Proceedings of the Thirty-First AAAI Conference on Artificial Intelligence (AAAI-17) Video Recovery via Learning Variation and Consistency of Images Zhouyuan Huo,1 Shangqian Gao,2 Weidong Cai,3 Heng Huang1∗ 1Department of Computer Science and Engineering, University of Texas at Arlington, USA 2College of Engineering, Northeastern University, USA 3School of Information Technologies, University of Sydney, NSW 2006, Australia [email protected], [email protected], [email protected], [email protected] Abstract 2010). However, trace norm is not the best approximation for rank minimization regularization. If a non-zero singular Matrix completion algorithms have been popularly used to recover images with missing entries, and they are proved value of a matrix varies, the trace norm value will change si- to be very effective. Recent works utilized tensor comple- multaneously, but the rank of the matrix keeps constant. So, tion models in video recovery assuming that all video frames there is a big gap between trace norm and rank minimiza- are homogeneous and correlated. However, real videos are tion, and a better model to approximate rank minimization made up of different episodes or scenes, i.e. heterogeneous. regularization is desired for learning a low-rank structure. Therefore, a video recovery model which utilizes both video In a recent work (Liu et al. 2009), Liu applied low-rank spatiotemporal consistency and variation is necessary. To model to solve image recovery problem. He assumed that solve this problem, we propose a new video recovery method video is in low-rank structure, however, videos are not ho- Sectional Trace Norm with Variation and Consistency Con- mogeneous, and they may contain sequences of images from straints (STN-VCC). -

Supporting Information for a Novel and Fast Method to Prepare Cu
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2020 Supporting Information for A novel and fast method to prepare Cu-supported α-Sb2S3@CuSbS2 binder-free electrode for sodium-ion batteries Jing Zhou a, Qirui Dou a, Lijuan Zhang a, Yingyu Wang a, Hao Yuan a, Jiangchun Chen a, b, Yu Cao *,a a School of Chemical Engineering, School of Electrical Engineering, Key Laboratory of Modern Power System Simulation and Control & Renewable Energy Technology, Ministry of Education, Northeast Electric Power University, Jilin 132012, China b School of Chemistry, Beijing Advanced Innovation Center for Biomedical Engineering, Beihang University, Beijing 100191, China Table S1. Formulations and corresponding active material loading of different kinds of electrodes. materials load of the Ratio of active mass of the Ref. electrode material, conductive active material carbon and binder -2 -2 Sb2S3 Hollow 1 mg cm 3:1:1 0.6 mg cm 1 Microspheres -2 -2 Sb2S3 1 mg cm 7:2:1 0.7 mg cm 2 -2 -2 Tin assisted Sb2S3 1.3 mg cm 7:2:1 0.91 mg cm 3 Nanoparticles -2 -2 Sb2S3/Reduced 1 mg cm 7:2:1 0.7 mg cm 4 Graphene Oxide -2 -2 Sb2S3 Nanosheets 1 mg cm 6:2:2 0.6 mg cm 5 -2 -2 Sb2S3/N-doped 0.8 mg cm 8:1:1 0.64 mg cm 6 carbon nanofiber Sb2Se3@NC@rGO 0.6-0.8 8:1:1 0.48-0.64 7 mg cm-2 mg cm-2 Our work 0.4-0.6 0.4-0.6 mg cm-2 mg cm-2 Table S2. -

Title Atmospheric Microplasma Based Binary Pt3co Nanoflowers Synthesis
Title Atmospheric microplasma based binary Pt3Co nanoflowers synthesis Author(s) Ying Wang, Bo Ouyang, Bowei Zhang, Yadian Boluo, Yizhong Huang, Raju V Ramanujan, Kostya (Ken) Ostrikov and Rajdeep S. Rawat Source Journal of Physics D: Applied Physics, 53(22), Article 225201 Published by IOP Publishing Copyright © 2020 IOP Publishing This is an author-created, un-copyedited version of an article accepted for publication in Journal of Physics D: Applied Physics. The publisher is not responsible for any errors or omissions in this version of the manuscript or any version derived from it. The Version of Record is available online at https://doi.org/10.1088/1361-6463/ab7797 Citation: Wang, Y., Ouyang, B., Zhang, B., Boluo, Y., Huang, Raju V. Ramanujan, Ostrikov, K. & Rawat, R. S. (2020). Atmospheric microplasma based binary Pt3Co nanoflowers synthesis. Journal of Physics D: Applied Physics, 53(22), Article 225201. https://doi.org/10.1088/1361-6463/ab7797 Atmospheric microplasma based binary Pt3Co nanoflowers synthesis Ying Wang,1,2,†,a) Bo Ouyang,1,2,† Bowei Zhang,3,4 Yadian Boluo,4 Yizhong Huang,4 R. V. Ramanujan,4 Kostya (Ken) Ostrikov5,6 and R. S. Rawat2,a) 1 School of Science, Nanjing University of Science and Technology, 200 Xiao Ling Wei Street, Nanjing, China 210094 2 National Science and Science Education (NSSE), National Institute of Education (NIE), Nanyang Technological University, 1 Nanyang Walk, Singapore 637616 3 Institute for Advanced Materials and Technology, University of Science and Technology Beijing, Beijing, China 100083 4 School of Material Science and Engineering (MSE), Nanyang Technological University, 50 Nanyang 8 Avenue, Singapore 639798 5 School of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology, Brisbane, QLD 4000, Australia 6 CSIRO-QUT Joint Sustainable Processes and Devices Laboratory, P.O. -

Curriculum Vitae
Curriculum Vitae BEILI WU The Stevens’ Lab Department of Molecular Biology The Scripps Research Institute, La Jolla, USA Telephone: 1-858-784-9411 (Lab), 1-858-366-8291 (Cell); E-mail: [email protected] PERSONAL INFORMATION First name: Beili Surname: Wu Sex: Female Date of birth: January 28th, 1979 Address: 3950 Mahaila Ave. APT. U13, San Diego, CA 92122, USA (home). The Scripps Research Institute, 10550 North Pines Road, GAC-1200, La Jolla, CA 92037, USA (Lab). EDUCATION AND TRAINING April, 2007 – present Research Associate Department of Molecular Biology, The Scripps Research Institute, La Jolla, USA September, 2001 - July, 2006 Ph.D. of Science Department of Biological Science and Biotechnology (September, 2001 – April, 2005) & Medical School (April, 2005 – July, 2006) Tsinghua University, Beijing, China September, 1997 - July, 2001 Bachelor of Science Department of Biology Beijing Normal University, Beijing, China AWARDS AND HONORS 2004 Huipu Scholarship, Tsinghua University 2002-2003 Guanghua Scholarship, Tsinghua University 2002 Lab Foundation and Contribution Scholarship, Tsinghua University 2001 Outstanding Graduate, Beijing Normal University; Outstanding Graduate, All the universities of Beijing 2000 Baogang Education Scholarship, Beijing Normal University 1999-2000 Second-Class Prize, Beijing Normal University 1998-1999 First-Class Prize, Beijing Normal University 1997-1998 Second-Class Prize, Beijing Normal University PUBLICATIONS 1. Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V & Stevens RC. Structures of the CXCR4 chemokine receptor in complex with small molecule and cyclic peptide antagonists. Science. (in press) 2. Wu B, Li P, Liu Y, Lou Z, Ding Y, Shu C, Ye S, Bartlam M, Shen B & Rao Z. -

Representing Talented Women in Eighteenth-Century Chinese Painting: Thirteen Female Disciples Seeking Instruction at the Lake Pavilion
REPRESENTING TALENTED WOMEN IN EIGHTEENTH-CENTURY CHINESE PAINTING: THIRTEEN FEMALE DISCIPLES SEEKING INSTRUCTION AT THE LAKE PAVILION By Copyright 2016 Janet C. Chen Submitted to the graduate degree program in Art History and the Graduate Faculty of the University of Kansas in partial fulfillment of the requirements for the degree of Doctor of Philosophy. ________________________________ Chairperson Marsha Haufler ________________________________ Amy McNair ________________________________ Sherry Fowler ________________________________ Jungsil Jenny Lee ________________________________ Keith McMahon Date Defended: May 13, 2016 The Dissertation Committee for Janet C. Chen certifies that this is the approved version of the following dissertation: REPRESENTING TALENTED WOMEN IN EIGHTEENTH-CENTURY CHINESE PAINTING: THIRTEEN FEMALE DISCIPLES SEEKING INSTRUCTION AT THE LAKE PAVILION ________________________________ Chairperson Marsha Haufler Date approved: May 13, 2016 ii Abstract As the first comprehensive art-historical study of the Qing poet Yuan Mei (1716–97) and the female intellectuals in his circle, this dissertation examines the depictions of these women in an eighteenth-century handscroll, Thirteen Female Disciples Seeking Instructions at the Lake Pavilion, related paintings, and the accompanying inscriptions. Created when an increasing number of women turned to the scholarly arts, in particular painting and poetry, these paintings documented the more receptive attitude of literati toward talented women and their support in the social and artistic lives of female intellectuals. These pictures show the women cultivating themselves through literati activities and poetic meditation in nature or gardens, common tropes in portraits of male scholars. The predominantly male patrons, painters, and colophon authors all took part in the formation of the women’s public identities as poets and artists; the first two determined the visual representations, and the third, through writings, confirmed and elaborated on the designated identities. -

Passages to Family History: a Guide to Genealogical Research in the Dartmouth College Library. PUB DATE 85 NOTE 106P.; Document Printed on Colored Paper
DOCUMENT RESUME ED 268 018 SO 016 975 AUTHOR Jaccaud, Robert D., Comp. TITLE Passages to Family History: A Guide to Genealogical Research in the Dartmouth College Library. PUB DATE 85 NOTE 106p.; Document printed on colored paper. PUB TYPE Historical Materials (060)-- Reference Materials - Bibliographies (131) EDRS PRICE MF01/PC05 Plus Postage. DESCRIPTORS Annotated Bibliographies; *Family History; *Genealogy; Higher Education; History; *Library Collections; Library Materials; Primary Sources; Publications; Reference Materials; Research Tools; Resource Materials IDENTIFIERS *Dartmouth College NH ABSTRACT Intended ' ;o assist the genealogist/family historian in locating and using resources inone of the oldest libraries in northern New England (Dartmouth College Library), this selective annotated bibliography contains over 300 citations relatedto family history and genealogical research. Followingan introdt.ctory section, materials are organized under nine subheadings: researchmaterials in the reference room; the library card catalog and online catalog; approaches to genealogical/family research in the subject catalog; research materials in the Baker Library Stacks;census data; selected collections and newspapers in the Jones Microtext Center;the Map Room; special collections in archives, manuscripts, andrare books; and selected research centers. Types of materials listed include menuals and aids; guides, indexes, and records; biographical information; heraldry, peerages, baronetages, gentry,etc.; bibliographies; indexes of names/genealogies; -

Chinese Romanization Table
239 Chinese Romanization Table Common Alphabetic (CA) follows the formula “consonants as in English, vowels as in Italian,” plus æ as in “cat,” v [compare the linguist’s !] as in “gut,” z as in “adz,” and yw (after l or n, simply w) for “umlaut u.” A lost initial ng- is restored to distinguish the states of We"! !! and Ngwe"! !!, both now “We"!.” Tones are h#!gh, r$!sing, lo%w, and fa"lling. The other systems are Pinyin (PY) and Wade-Giles (WG). CA PY WG CA PY WG a a a chya qia ch!ia ai ai ai chyang qiang ch!iang an an an chyau qiao ch!iao ang ang ang chye qie ch!ieh ar er erh chyen qian ch!ien au ao ao chyou qiu ch!iu ba ba pa chyung qiong ch!iung bai bai pai chyw qu ch!ü ban ban pan chywæn quan ch!üan bang bang pang chywe que ch!üeh bau bao pao chywn qun ch!ün bei bei pei da da ta bi bi pi dai dai tai bin bin pin dan dan tan bing bing ping dang dang tang bu bu pu dau dao tao bvn ben pen dei dei tei bvng beng peng di di ti bwo bo po ding ding ting byau biao piao dou dou tou bye bie pieh du du tu byen bian pien dun dun tun cha cha ch!a dung dong tung chai chai ch!ai dv de te chan chan ch!an dvng deng teng chang chang ch!ang dwan duan tuan chau chao ch!ao dwei dui tui chi qi ch!i dwo duo to chin qin ch!in dyau diao tiao ching qing ch!ing dye die tieh chou chou ch!ou dyen dian tien chr chi ch!ih dyou diu tiu chu chu ch!u dz zi tzu chun chun ch!un dza za tsa chung chong ch!ung dzai zai tsai chv che ch!e dzan zan tsan chvn chen ch!en dzang zang tsang chvng cheng ch!eng dzau zao tsao chwai chuai ch!uai dzei zei tsei chwan chuan ch!uan dzou