Methods of Analysis by the U.S. Geological Survey National Water
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lifetime Organophosphorous Insecticide Use Among Private Pesticide Applicators in the Agricultural Health Study
Journal of Exposure Science and Environmental Epidemiology (2012) 22, 584 -- 592 & 2012 Nature America, Inc. All rights reserved 1559-0631/12 www.nature.com/jes ORIGINAL ARTICLE Lifetime organophosphorous insecticide use among private pesticide applicators in the Agricultural Health Study Jane A. Hoppin1, Stuart Long2, David M. Umbach3, Jay H. Lubin4, Sarah E. Starks5, Fred Gerr5, Kent Thomas6, Cynthia J. Hines7, Scott Weichenthal8, Freya Kamel1, Stella Koutros9, Michael Alavanja9, Laura E. Beane Freeman9 and Dale P. Sandler1 Organophosphorous insecticides (OPs) are the most commonly used insecticides in US agriculture, but little information is available regarding specific OP use by individual farmers. We describe OP use for licensed private pesticide applicators from Iowa and North Carolina in the Agricultural Health Study (AHS) using lifetime pesticide use data from 701 randomly selected male participants collected at three time periods. Of 27 OPs studied, 20 were used by 41%. Overall, 95% had ever applied at least one OP. The median number of different OPs used was 4 (maximum ¼ 13). Malathion was the most commonly used OP (74%) followed by chlorpyrifos (54%). OP use declined over time. At the first interview (1993--1997), 68% of participants had applied OPs in the past year; by the last interview (2005--2007), only 42% had. Similarly, median annual application days of OPs declined from 13.5 to 6 days. Although OP use was common, the specific OPs used varied by state, time period, and individual. Much of the variability in OP use was associated with the choice of OP, rather than the frequency or duration of application. -
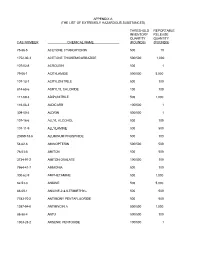
The List of Extremely Hazardous Substances)
APPENDIX A (THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES) THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 75-86-5 ACETONE CYANOHYDRIN 500 10 1752-30-3 ACETONE THIOSEMICARBAZIDE 500/500 1,000 107-02-8 ACROLEIN 500 1 79-06-1 ACRYLAMIDE 500/500 5,000 107-13-1 ACRYLONITRILE 500 100 814-68-6 ACRYLYL CHLORIDE 100 100 111-69-3 ADIPONITRILE 500 1,000 116-06-3 ALDICARB 100/500 1 309-00-2 ALDRIN 500/500 1 107-18-6 ALLYL ALCOHOL 500 100 107-11-9 ALLYLAMINE 500 500 20859-73-8 ALUMINUM PHOSPHIDE 500 100 54-62-6 AMINOPTERIN 500/500 500 78-53-5 AMITON 500 500 3734-97-2 AMITON OXALATE 100/500 100 7664-41-7 AMMONIA 500 100 300-62-9 AMPHETAMINE 500 1,000 62-53-3 ANILINE 500 5,000 88-05-1 ANILINE,2,4,6-TRIMETHYL- 500 500 7783-70-2 ANTIMONY PENTAFLUORIDE 500 500 1397-94-0 ANTIMYCIN A 500/500 1,000 86-88-4 ANTU 500/500 100 1303-28-2 ARSENIC PENTOXIDE 100/500 1 THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 1327-53-3 ARSENOUS OXIDE 100/500 1 7784-34-1 ARSENOUS TRICHLORIDE 500 1 7784-42-1 ARSINE 100 100 2642-71-9 AZINPHOS-ETHYL 100/500 100 86-50-0 AZINPHOS-METHYL 10/500 1 98-87-3 BENZAL CHLORIDE 500 5,000 98-16-8 BENZENAMINE, 3-(TRIFLUOROMETHYL)- 500 500 100-14-1 BENZENE, 1-(CHLOROMETHYL)-4-NITRO- 500/500 500 98-05-5 BENZENEARSONIC ACID 10/500 10 3615-21-2 BENZIMIDAZOLE, 4,5-DICHLORO-2-(TRI- 500/500 500 FLUOROMETHYL)- 98-07-7 BENZOTRICHLORIDE 100 10 100-44-7 BENZYL CHLORIDE 500 100 140-29-4 BENZYL CYANIDE 500 500 15271-41-7 BICYCLO[2.2.1]HEPTANE-2-CARBONITRILE,5- -

744 Hydrolysis of Chiral Organophosphorus Compounds By
[Frontiers in Bioscience, Landmark, 26, 744-770, Jan 1, 2021] Hydrolysis of chiral organophosphorus compounds by phosphotriesterases and mammalian paraoxonase-1 Antonio Monroy-Noyola1, Damianys Almenares-Lopez2, Eugenio Vilanova Gisbert3 1Laboratorio de Neuroproteccion, Facultad de Farmacia, Universidad Autonoma del Estado de Morelos, Morelos, Mexico, 2Division de Ciencias Basicas e Ingenierias, Universidad Popular de la Chontalpa, H. Cardenas, Tabasco, Mexico, 3Instituto de Bioingenieria, Universidad Miguel Hernandez, Elche, Alicante, Spain TABLE OF CONTENTS 1. Abstract 2. Introduction 2.1. Organophosphorus compounds (OPs) and their toxicity 2.2. Metabolism and treatment of OP intoxication 2.3. Chiral OPs 3. Stereoselective hydrolysis 3.1. Stereoselective hydrolysis determines the toxicity of chiral compounds 3.2. Hydrolysis of nerve agents by PTEs 3.2.1. Hydrolysis of V-type agents 3.3. PON1, a protein restricted in its ability to hydrolyze chiral OPs 3.4. Toxicity and stereoselective hydrolysis of OPs in animal tissues 3.4.1. The calcium-dependent stereoselective activity of OPs associated with PON1 3.4.2. Stereoselective hydrolysis commercial OPs pesticides by alloforms of PON1 Q192R 3.4.3. PON1, an enzyme that stereoselectively hydrolyzes OP nerve agents 3.4.4. PON1 recombinants and stereoselective hydrolysis of OP nerve agents 3.5. The activity of PTEs in birds 4. Conclusions 5. Acknowledgments 6. References 1. ABSTRACT Some organophosphorus compounds interaction of the racemic OPs with these B- (OPs), which are used in the manufacturing of esterases (AChE and NTE) and such interactions insecticides and nerve agents, are racemic mixtures have been studied in vivo, ex vivo and in vitro, using with at least one chiral center with a phosphorus stereoselective hydrolysis by A-esterases or atom. -

Acutely / Extremely Hazardous Waste List
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extemely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extemely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extemely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extemely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extemely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extemely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extemely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extemely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extemely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Soil Degradation Comparisons of Corn Rootworm Insecticides in the Field and Laboratory
South Dakota State University Open PRAIRIE: Open Public Research Access Institutional Repository and Information Exchange Electronic Theses and Dissertations 1987 Soil Degradation Comparisons of Corn Rootworm Insecticides in the Field and Laboratory Michael J. Haas Follow this and additional works at: https://openprairie.sdstate.edu/etd Recommended Citation Haas, Michael J., "Soil Degradation Comparisons of Corn Rootworm Insecticides in the Field and Laboratory" (1987). Electronic Theses and Dissertations. 4447. https://openprairie.sdstate.edu/etd/4447 This Thesis - Open Access is brought to you for free and open access by Open PRAIRIE: Open Public Research Access Institutional Repository and Information Exchange. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Open PRAIRIE: Open Public Research Access Institutional Repository and Information Exchange. For more information, please contact [email protected]. SOIL DEGRADATION COMPARISONS OF CORN ROOTWORM INSECTICIDES IN THE FIELD AND LABORATORY BY MICHAEL J . HAAS A thesis submitted in partial fulfillment of the requirements for the degree Master of Science Major in Entomo logy South Dakota State Univers ity 1987 SOIL DEGRADATION COMPARISONS OF INSECTICIDES IN THE FIELD AND LA BORATORY This thesis is approved as a creditable and independent investigation by a cand idate for the degree , Master of Science , and is acceptable for meeting the thesis requirements for this degree . Acceptance of this thesis does not imply that the conclusions reached by the candidate are necessarily the conclusions of the major department . David D. Walgenbach Thesis Adviser j MaJrlce L. Horton Date �ead , Dept . of Plant Science ACKNOWLEDGEMENTS I would like to extend my sincere gratitude to Dr . -

Environmental Health Criteria 63 ORGANOPHOSPHORUS
Environmental Health Criteria 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION Please note that the layout and pagination of this web version are not identical with the printed version. Organophophorus insecticides: a general introduction (EHC 63, 1986) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1986 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, coordination -
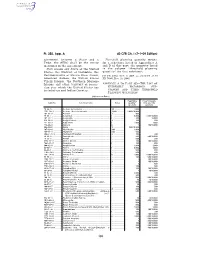
40 CFR Ch. I (7–1–09 Edition) Pt. 355, App. A
Pt. 355, App. A 40 CFR Ch. I (7–1–09 Edition) agreement between a State and a Threshold planning quantity means, Tribe, the SERC shall be the entity for a substance listed in Appendices A identified in the agreement. and B of this part, the quantity listed State means any State of the United in the column ‘‘threshold planning States, the District of Columbia, the quantity’’ for that substance. Commonwealth of Puerto Rico, Guam, [73 FR 65462, Nov. 3, 2008, as amended at 73 American Samoa, the United States FR 76960, Dec. 18, 2008] Virgin Islands, the Northern Mariana Islands, any other territory or posses- APPENDIX A TO PART 355—THE LIST OF sion over which the United States has EXTREMELY HAZARDOUS SUB- jurisdiction and Indian Country. STANCES AND THEIR THRESHOLD PLANNING QUANTITIES [Alphabetical Order] Reportable Threshold plan- CAS No. Chemical name Notes quantity * ning quantity (pounds) (pounds) 75–86–5 ................ Acetone Cyanohydrin ................................................. 10 ................ 1,000 1752–30–3 ............ Acetone Thiosemicarbazide ....................................... 1,000 ........... 1,000/10,000 107–02–8 .............. Acrolein ....................................................................... 1 .................. 500 79–06–1 ................ Acrylamide .................................................................. f ................... 5,000 1,000/10,000 107–13–1 .............. Acrylonitrile ................................................................. f ................... 100 10,000 -

Insecticides
fY)I\) -;; ooo 3tfJ INSECTICIDES Extension Bulletin 387-Revlsed 1980 AGAfCULTURAL EXTENSION SERVICE UNIVERSITY OF MINNESOTA Contents General precautions for using pesticides . 4 Safety precautions and first aid . 4 Minnesota poison information centers . 5 Protecting honey bees from insecticides . 6 Pesticide toxicity and LD 50's . • . • . 6 Acute oral and dermal LD 50's for insecticides . 7 Forms of insecticides . 8 Calculating dosage and rates of application . 9 Sprayer calibration . 11 Description of insecticides, miticides ........................... 12 Chlorinated hydrocarbons ................................. 12 Carbamates .............................................. 13 Organophosphates ........................................ 14 Sulfonates, carbonates, botanicals, and miscellaneous groups ... 18 Legal Restrictions on Use of Pesticides The Federal Insecticide, Fungicide and Rodenticide Act and the Minnesota Pesticide Act of 1976, require that those who use or supervise the use of certain pesticides with restricted uses must be certified. The labels of those pesticides with restricted uses will contain information regarding these restrictions. Be sure to read all labels thoroughly and use any pesticide for the crops and pests listed on the label only. Information about applicator certification may be obtained from your County Extension Director or the Minnesota Depmt ment of Agriculture. The U.S. Environmental Protection Agency (EPA) has designated the following pesticides for reshicted use: acrolein endrin mevinphos (Phosdrin) acrylonitrile ethyl parathion paraquat aldicarb (Temik) 1080 piclorarri (Tordon) allyl alcohol 1081 sodium cyanide alluminum phosphide (Phostoxin) hydrocyanic acid strychnine azinphos methyl (Guthion) methomyl (Lannate, Nudrin) sulfotepp calcium cyanide methyl bromide tepp demeton (Systox) methyl parathion Authors of this publication are J. A. Lofgren, professor and extension entomologist; D. M. Noetzel, assistant professor and extension entomologist; P. K. Hareln, professor and extension entomologist; M. -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals -

Environmental Protection Agency Pt. 355, App. B
Environmental Protection Agency Pt. 355, App. B [Alphabetical Order] Reportable Threshold plan- CAS No. Chemical name Notes quantity * ning quantity (pounds) (pounds) 5344–82–1 ............ Thiourea, (2-Chlorophenyl)- ....................................... ..................... 100 100/10,000 614–78–8 .............. Thiourea, (2-Methylphenyl)- ....................................... ..................... 500 500/10,000 7550–45–0 ............ Titanium Tetrachloride ................................................ ..................... 1,000 100 584–84–9 .............. Toluene 2,4-Diisocyanate ........................................... ..................... 100 500 91–08–7 ................ Toluene 2,6-Diisocyanate ........................................... ..................... 100 100 110–57–6 .............. Trans-1,4-Dichlorobutene ........................................... ..................... 500 500 1031–47–6 ............ Triamiphos .................................................................. ..................... 500 500/10,000 24017–47–8 .......... Triazofos ..................................................................... ..................... 500 500 76–02–8 ................ Trichloroacetyl Chloride .............................................. ..................... 500 500 115–21–9 .............. Trichloroethylsilane ..................................................... d .................. 500 500 327–98–0 .............. Trichloronate ............................................................... e ................. -

Fonofos Method No.: PV2027 Target Concentration: 0.1 Mg/M3 Skin
Fonofos Method no.: PV2027 Target Concentration: 0.1 mg/m3 Skin (ACGIH TLV-TWA) Procedure: Samples are collected by drawing a known volume of air through an OSHA versatile sampler (OVS-2) tube, containing a glass fiber filter and two sections of XAD-2 adsorbent. Samples are desorbed with toluene and an internal standard and analyzed by gas chromatography (GC) using a flame photometric detector (FPD) in the phosphorus mode. Recommended air volume and sampling rate: 480 minutes at 1.0 L/min (480 L) Detection limit of the overall procedure 0.00044 mg/m3 (based on the recommended air volume and the analytical detection limit): Status of method: Partially Validated method. This method has been partially evaluated and is presented for information and trial use only. July 1989 (final) David B. Armitage Carcinogen and Pesticide Branch OSHA Salt Lake Technical Center Sandy UT 84070 1 of 8 1 General Discussion 1.1 Background 1.1.1 History of procedure This evaluation was undertaken to determine the effectiveness of the OVS-2 tube as a sampling device for fonofos. It follows the procedure developed for several other organophosphorus pesticides. (Ref. 5.1) 1.1.2 Toxic effects (This section is for information only and should not be taken as the basis of OSHA policy). The following paragraph is excerpted from the book OCCUPATIONAL DISEASES, A Guide to Their Recognition. (Ref. 5.2) The organic phosphorus compounds act as irreversible inhibitors of cholinesterase, thereby allowing the accumulation of large amounts of acetylcholine. When a critical level of cholinesterase depletion is reached, usually about 20% of normal, symptoms, and signs of acetylcholine accumulation poisoning become manifest. -

Seasonal Pesticide Use in a Rural Community on the US/Mexico Border
Journal of Exposure Analysis and Environmental Epidemiology (2004) 14, 473–478 r 2004 Nature Publishing Group All rights reserved 1053-4245/04/$30.00 www.nature.com/jea Seasonal pesticide use in a rural community on the US/Mexico border NATALIE C.G. FREEMAN,a STUART L. SHALAT,a KATHLEEN BLACK,a MARTA JIMENEZ,a KIRBY C. DONNELLY,b A. CALVINb ANDJUANRAMIREZc aEnvironmental and Occupational Health Sciences Institute, a jointly sponsored institute of Rutgers the State University of New Jersey and University of Medicine & Dentistry of New Jersey F Robert Wood Johnson Medical School, Piscataway, NJ, USA bCenter for Environmental and Rural Health, Texas A&MUniversity, College Station, TX, USA cTDI-Brooks International, College Station, TX, USA An environmental measurement and correlation study of infant and toddler exposure to pesticides was carried out in a colonia south of the city of Laredo, Texas. As part of the study, homes were visited during the late spring or summer, and during the winter of 2000–2001. At each visit, families reported on their use of pesticides in and around the home and floor wipe samples were collected and analyzed for 14 organophosphate and triazine pesticides. Selection of homes was based on the presence of infants and toddlers. A total of 27 homes participated in both seasonal visits. The interval between visits was 671.4 months. Univariate and multivariate nonparametric analyses were carried out using SPSSs statistical software. Pesticide use within the home was more often reported than outside use and showed seasonal variation in use patterns. Indoor use was primarily associated with ants and cockroaches, and secondarily with rodents.