Estimating Causal Effects
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Descriptive Statistics (Part 2): Interpreting Study Results
Statistical Notes II Descriptive statistics (Part 2): Interpreting study results A Cook and A Sheikh he previous paper in this series looked at ‘baseline’. Investigations of treatment effects can be descriptive statistics, showing how to use and made in similar fashion by comparisons of disease T interpret fundamental measures such as the probability in treated and untreated patients. mean and standard deviation. Here we continue with descriptive statistics, looking at measures more The relative risk (RR), also sometimes known as specific to medical research. We start by defining the risk ratio, compares the risk of exposed and risk and odds, the two basic measures of disease unexposed subjects, while the odds ratio (OR) probability. Then we show how the effect of a disease compares odds. A relative risk or odds ratio greater risk factor, or a treatment, can be measured using the than one indicates an exposure to be harmful, while relative risk or the odds ratio. Finally we discuss the a value less than one indicates a protective effect. ‘number needed to treat’, a measure derived from the RR = 1.2 means exposed people are 20% more likely relative risk, which has gained popularity because of to be diseased, RR = 1.4 means 40% more likely. its clinical usefulness. Examples from the literature OR = 1.2 means that the odds of disease is 20% higher are used to illustrate important concepts. in exposed people. RISK AND ODDS Among workers at factory two (‘exposed’ workers) The probability of an individual becoming diseased the risk is 13 / 116 = 0.11, compared to an ‘unexposed’ is the risk. -

Meta-Analysis of Proportions
NCSS Statistical Software NCSS.com Chapter 456 Meta-Analysis of Proportions Introduction This module performs a meta-analysis of a set of two-group, binary-event studies. These studies have a treatment group (arm) and a control group. The results of each study may be summarized as counts in a 2-by-2 table. The program provides a complete set of numeric reports and plots to allow the investigation and presentation of the studies. The plots include the forest plot, radial plot, and L’Abbe plot. Both fixed-, and random-, effects models are available for analysis. Meta-Analysis refers to methods for the systematic review of a set of individual studies with the aim to combine their results. Meta-analysis has become popular for a number of reasons: 1. The adoption of evidence-based medicine which requires that all reliable information is considered. 2. The desire to avoid narrative reviews which are often misleading. 3. The desire to interpret the large number of studies that may have been conducted about a specific treatment. 4. The desire to increase the statistical power of the results be combining many small-size studies. The goals of meta-analysis may be summarized as follows. A meta-analysis seeks to systematically review all pertinent evidence, provide quantitative summaries, integrate results across studies, and provide an overall interpretation of these studies. We have found many books and articles on meta-analysis. In this chapter, we briefly summarize the information in Sutton et al (2000) and Thompson (1998). Refer to those sources for more details about how to conduct a meta- analysis. -

Philosophy of Science and Philosophy of Chemistry
Philosophy of Science and Philosophy of Chemistry Jaap van Brakel Abstract: In this paper I assess the relation between philosophy of chemistry and (general) philosophy of science, focusing on those themes in the philoso- phy of chemistry that may bring about major revisions or extensions of cur- rent philosophy of science. Three themes can claim to make a unique contri- bution to philosophy of science: first, the variety of materials in the (natural and artificial) world; second, extending the world by making new stuff; and, third, specific features of the relations between chemistry and physics. Keywords : philosophy of science, philosophy of chemistry, interdiscourse relations, making stuff, variety of substances . 1. Introduction Chemistry is unique and distinguishes itself from all other sciences, with respect to three broad issues: • A (variety of) stuff perspective, requiring conceptual analysis of the notion of stuff or material (Sections 4 and 5). • A making stuff perspective: the transformation of stuff by chemical reaction or phase transition (Section 6). • The pivotal role of the relations between chemistry and physics in connection with the question how everything fits together (Section 7). All themes in the philosophy of chemistry can be classified in one of these three clusters or make contributions to general philosophy of science that, as yet , are not particularly different from similar contributions from other sci- ences (Section 3). I do not exclude the possibility of there being more than three clusters of philosophical issues unique to philosophy of chemistry, but I am not aware of any as yet. Moreover, highlighting the issues discussed in Sections 5-7 does not mean that issues reviewed in Section 3 are less im- portant in revising the philosophy of science. -

Causal Inference with Information Fields
Causal Inference with Information Fields Benjamin Heymann Michel De Lara Criteo AI Lab, Paris, France CERMICS, École des Ponts, Marne-la-Vallée, France [email protected] [email protected] Jean-Philippe Chancelier CERMICS, École des Ponts, Marne-la-Vallée, France [email protected] Abstract Inferring the potential consequences of an unobserved event is a fundamental scientific question. To this end, Pearl’s celebrated do-calculus provides a set of inference rules to derive an interventional probability from an observational one. In this framework, the primitive causal relations are encoded as functional dependencies in a Structural Causal Model (SCM), which maps into a Directed Acyclic Graph (DAG) in the absence of cycles. In this paper, by contrast, we capture causality without reference to graphs or functional dependencies, but with information fields. The three rules of do-calculus reduce to a unique sufficient condition for conditional independence: the topological separation, which presents some theoretical and practical advantages over the d-separation. With this unique rule, we can deal with systems that cannot be represented with DAGs, for instance systems with cycles and/or ‘spurious’ edges. We provide an example that cannot be handled – to the extent of our knowledge – with the tools of the current literature. 1 Introduction As the world shifts toward more and more data-driven decision-making, causal inference is taking more space in applied sciences, statistics and machine learning. This is because it allows for better, more robust decision-making, and provides a way to interpret the data that goes beyond correlation Pearl and Mackenzie [2018]. -
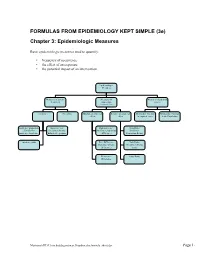
FORMULAS from EPIDEMIOLOGY KEPT SIMPLE (3E) Chapter 3: Epidemiologic Measures
FORMULAS FROM EPIDEMIOLOGY KEPT SIMPLE (3e) Chapter 3: Epidemiologic Measures Basic epidemiologic measures used to quantify: • frequency of occurrence • the effect of an exposure • the potential impact of an intervention. Epidemiologic Measures Measures of disease Measures of Measures of potential frequency association impact (“Measures of Effect”) Incidence Prevalence Absolute measures of Relative measures of Attributable Fraction Attributable Fraction effect effect in exposed cases in the Population Incidence proportion Incidence rate Risk difference Risk Ratio (Cumulative (incidence density, (Incidence proportion (Incidence Incidence, Incidence hazard rate, person- difference) Proportion Ratio) Risk) time rate) Incidence odds Rate Difference Rate Ratio (Incidence density (Incidence density difference) ratio) Prevalence Odds Ratio Difference Macintosh HD:Users:buddygerstman:Dropbox:eks:formula_sheet.doc Page 1 of 7 3.1 Measures of Disease Frequency No. of onsets Incidence Proportion = No. at risk at beginning of follow-up • Also called risk, average risk, and cumulative incidence. • Can be measured in cohorts (closed populations) only. • Requires follow-up of individuals. No. of onsets Incidence Rate = ∑person-time • Also called incidence density and average hazard. • When disease is rare (incidence proportion < 5%), incidence rate ≈ incidence proportion. • In cohorts (closed populations), it is best to sum individual person-time longitudinally. It can also be estimated as Σperson-time ≈ (average population size) × (duration of follow-up). Actuarial adjustments may be needed when the disease outcome is not rare. • In an open populations, Σperson-time ≈ (average population size) × (duration of follow-up). Examples of incidence rates in open populations include: births Crude birth rate (per m) = × m mid-year population size deaths Crude mortality rate (per m) = × m mid-year population size deaths < 1 year of age Infant mortality rate (per m) = × m live births No. -

The Practice of Causal Inference in Cancer Epidemiology
Vol. 5. 303-31 1, April 1996 Cancer Epidemiology, Biomarkers & Prevention 303 Review The Practice of Causal Inference in Cancer Epidemiology Douglas L. Weedt and Lester S. Gorelic causes lung cancer (3) and to guide causal inference for occu- Preventive Oncology Branch ID. L. W.l and Comprehensive Minority pational and environmental diseases (4). From 1965 to 1995, Biomedical Program IL. S. 0.1. National Cancer Institute, Bethesda, Maryland many associations have been examined in terms of the central 20892 questions of causal inference. Causal inference is often practiced in review articles and editorials. There, epidemiologists (and others) summarize evi- Abstract dence and consider the issues of causality and public health Causal inference is an important link between the recommendations for specific exposure-cancer associations. practice of cancer epidemiology and effective cancer The purpose of this paper is to take a first step toward system- prevention. Although many papers and epidemiology atically reviewing the practice of causal inference in cancer textbooks have vigorously debated theoretical issues in epidemiology. Techniques used to assess causation and to make causal inference, almost no attention has been paid to the public health recommendations are summarized for two asso- issue of how causal inference is practiced. In this paper, ciations: alcohol and breast cancer, and vasectomy and prostate we review two series of review papers published between cancer. The alcohol and breast cancer association is timely, 1985 and 1994 to find answers to the following questions: controversial, and in the public eye (5). It involves a common which studies and prior review papers were cited, which exposure and a common cancer and has a large body of em- causal criteria were used, and what causal conclusions pirical evidence; over 50 studies and over a dozen reviews have and public health recommendations ensued. -

Statistics and Causal Inference (With Discussion)
Applied Statistics Lecture Notes Kosuke Imai Department of Politics Princeton University February 2, 2008 Making statistical inferences means to learn about what you do not observe, which is called parameters, from what you do observe, which is called data. We learn the basic principles of statistical inference from a perspective of causal inference, which is a popular goal of political science research. Namely, we study statistics by learning how to make causal inferences with statistical methods. 1 Statistical Framework of Causal Inference What do we exactly mean when we say “An event A causes another event B”? Whether explicitly or implicitly, this question is asked and answered all the time in political science research. The most commonly used statistical framework of causality is based on the notion of counterfactuals (see Holland, 1986). That is, we ask the question “What would have happened if an event A were absent (or existent)?” The following example illustrates the fact that some causal questions are more difficult to answer than others. Example 1 (Counterfactual and Causality) Interpret each of the following statements as a causal statement. 1. A politician voted for the education bill because she is a democrat. 2. A politician voted for the education bill because she is liberal. 3. A politician voted for the education bill because she is a woman. In this framework, therefore, the fundamental problem of causal inference is that the coun- terfactual outcomes cannot be observed, and yet any causal inference requires both factual and counterfactual outcomes. This idea is formalized below using the potential outcomes notation. -

Bayesian Causal Inference
Bayesian Causal Inference Maximilian Kurthen Master’s Thesis Max Planck Institute for Astrophysics Within the Elite Master Program Theoretical and Mathematical Physics Ludwig Maximilian University of Munich Technical University of Munich Supervisor: PD Dr. Torsten Enßlin Munich, September 12, 2018 Abstract In this thesis we address the problem of two-variable causal inference. This task refers to inferring an existing causal relation between two random variables (i.e. X → Y or Y → X ) from purely observational data. We begin by outlining a few basic definitions in the context of causal discovery, following the widely used do-Calculus [Pea00]. We continue by briefly reviewing a number of state-of-the-art methods, including very recent ones such as CGNN [Gou+17] and KCDC [MST18]. The main contribution is the introduction of a novel inference model where we assume a Bayesian hierarchical model, pursuing the strategy of Bayesian model selection. In our model the distribution of the cause variable is given by a Poisson lognormal distribution, which allows to explicitly regard discretization effects. We assume Fourier diagonal covariance operators, where the values on the diagonal are given by power spectra. In the most shallow model these power spectra and the noise variance are fixed hyperparameters. In a deeper inference model we replace the noise variance as a given prior by expanding the inference over the noise variance itself, assuming only a smooth spatial structure of the noise variance. Finally, we make a similar expansion for the power spectra, replacing fixed power spectra as hyperparameters by an inference over those, where again smoothness enforcing priors are assumed. -

Experience Verification Form
Tennessee Department of Education – Office of Educator Licensure Experience Verification Form The information listed below is to be completed by the current or previous employer (Superintendent, Headmaster, Agency Director, or Designated Personnel Officer). Principals are not authorized to sign this form unless they are the designated personnel officer. Use one line for each change in status. Do not include leave of absence periods. Only include experience that is not in the TN state data system. Name of Educator: ________________________________________________ Social Security Number: _________________________ Information below to be completed by the SCHOOL SYSTEM where the teaching was performed. Experience Record: Please list experience yearly, each year on a separate line, beginning with July 1 and ending June 30. Name School Position Fiscal Year, July 01 - June 30 Time Employed % Time, Total of System and State Beginning Date Ending Date Months / Days Ex. Part-time, Days in Month/Day/Year Month/Day/Year Full-time School Year School Grade Level Per Year Check one of the following: Public School Private School * Charter School * U.S. Government Service Teaching Program * ** Please note: If non-public school you must identify accreditation. The above school/school system was fully approved or accredited by the ____________________________________________________________________ at the time the service was performed. (State Department of Education, or Regional Association of Colleges & Schools, or recognized private school accrediting -

C.S. Peirce's Abduction from the Prior Analytics
Providence College DigitalCommons@Providence Community Scholar Publications Providence College Community Scholars 7-1996 C.S. Peirce's Abduction from the Prior Analytics William Paul Haas Follow this and additional works at: https://digitalcommons.providence.edu/comm_scholar_pubs Haas, William Paul, "C.S. Peirce's Abduction from the Prior Analytics" (1996). Community Scholar Publications. 2. https://digitalcommons.providence.edu/comm_scholar_pubs/2 This Article is brought to you for free and open access by the Providence College Community Scholars at DigitalCommons@Providence. It has been accepted for inclusion in Community Scholar Publications by an authorized administrator of DigitalCommons@Providence. For more information, please contact [email protected]. WILLIAM PAUL HAAS July 1996 C.S. PEIRCE'S ABDUCTION FROM THE PRIOR ANALYTICS In his Ancient Formal Logic. Professor Joseph Bochenski finds Aristotle's description of syllogisms based upon hypotheses to be "difficult to understand." Noting that we do not have the treatise which Aristotle promised to write, Bochenski laments the fact that the Prior Analytics, where it is treated most explicitly, "is either corrupted or (which is more probable) was hastily written and contains logical errors." (1) Charles Sanders Peirce wrestled with the same difficult text when he attempted to establish the Aristotelian roots of his theory of abductive or hypothetical reasoning. However, Peirce opted for the explanation that the fault was with the corrupted text, not with Aristotle's exposition. Peirce interpreted the text of Book II, Chapter 25 thus: Accordingly, when he opens the next chapter with the word ' Ajray (¿y-q a word evidently chosen to form a pendant to 'Errctyuyrj, we feel sure that this is what he is coming to. -

Changes to the Standard to Implement the Safer Choice Label (Pdf)
Changes to the Standard to Implement the Safer Choice Label 2/10/15 Implementation of a New Label for the DfE Safer Product Labeling Program and Supporting Modifications to the DfE Standard for Safer Products, now to be known as the “Safer Choice Standard,” including the Designation of a Fragrance-Free Label This document explains several important changes to EPA’s Safer Product Labeling Program (SPLP), as well as a number of conforming changes to the program’s Standard for Safer Products, including: a new label1 and name for the EPA SPLP; an associated fragrance-free label; and related changes to the standard that qualifies products for the label. With this document EPA is finalizing the new label for immediate use. While effective immediately, the Agency is requesting comment on the other changes described in this document, including a new, optional fragrance-free certification. Redesigned Label The EPA Design for the Environment (DfE) Safer Product Labeling Program has redesigned its label to better communicate the program’s human health and environmental protection goals, increase consumer and institutional/industrial purchaser understanding and recognition of products bearing the label, and encourage innovation and the development of safer chemicals and chemical-based products. The label’s new name “Safer Choice,” with the accompanying wording (or “tagline”) “Meets U.S. EPA Safer Product Standards” (which is part of the label), and design were developed over many months based on input and feedback from a diverse set of stakeholders and the general public. This input and feedback included more than 1700 comments on EPA’s website, six consumer focus group sessions in three geographic markets, and a national online survey. -

Articles Causal Inference in Civil Rights Litigation
VOLUME 122 DECEMBER 2008 NUMBER 2 © 2008 by The Harvard Law Review Association ARTICLES CAUSAL INFERENCE IN CIVIL RIGHTS LITIGATION D. James Greiner TABLE OF CONTENTS I. REGRESSION’S DIFFICULTIES AND THE ABSENCE OF A CAUSAL INFERENCE FRAMEWORK ...................................................540 A. What Is Regression? .........................................................................................................540 B. Problems with Regression: Bias, Ill-Posed Questions, and Specification Issues .....543 1. Bias of the Analyst......................................................................................................544 2. Ill-Posed Questions......................................................................................................544 3. Nature of the Model ...................................................................................................545 4. One Regression, or Two?............................................................................................555 C. The Fundamental Problem: Absence of a Causal Framework ....................................556 II. POTENTIAL OUTCOMES: DEFINING CAUSAL EFFECTS AND BEYOND .................557 A. Primitive Concepts: Treatment, Units, and the Fundamental Problem.....................558 B. Additional Units and the Non-Interference Assumption.............................................560 C. Donating Values: Filling in the Missing Counterfactuals ...........................................562 D. Randomization: Balance in Background Variables ......................................................563