Porous Drug Matrices and Methods of Manufacture
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

United States Patent Office Patented May 21, 1957 1
2,793,218 United States Patent Office Patented May 21, 1957 1. 2,793,218 9-HALO-11.KETO.17-ALKYTLTESTOSTERONES Milton E. Herr, Kalamazoo, Mich., assignor to The Up john Company, Kalamazoo, Mich., a corporation of Michigan No Drawing. Original application August 8, 1955, Serial No. 527,118. Divided and this application May 10, WII 1956, Serial No. 583,922 wherein R is a lower-alkyl group containing less than three carbon atoms, i. e., methyl or ethyl; R is hydrogen 3 Claims. (Cl. 260-397.45) or the acyl radical of a hydrocarbon carboxylic acid con taining from one to twelve carbon atoms, inclusive; X This invention relates to novel 17-alkyl-17-hydroxy 5 is a halogen having an atomic weight from 79 to 127, in steroids and esters thereof. clusive, i. e., bromine or iodine, X is a halogen having It is an object of this invention to provide novel 9a an atomic weight from 19 to 36, inclusive, i. e., fluorine halo - 116 - hydroxy - 17 - alkyltestosterones, 9a - halo or chlorine, and X' is a halogen having an atomic weight 11 - keto - 17 - alkyltestosterones, 17-esters thereof, and from 19 to 127, inclusive, i. e., fluorine, chlorine, bromine intermediates in the production thereof. Another object 20 or iodine. is the provision of processes for the production thereof. Following the series of reactions described hereinafter Other objects will be apparent to those skilled in the art for the conversion of 11o - hydroxy - 17 - methyltestos to which this invention pertains. terone (I) to 9a - hydroxy - 17 - methyltestosterone According to the present invention, the novel 9a-halo testosterone and esters thereof (VII), but substituting 10 116 - hydroxy - 17 - alkyltestosterones, 9a - halo - 11 25 normethyl - 11a - hydroxy - 17 - methyltestosterone keto - 17 - alkyltestosterones and 17 - esters thereof may (U. -

Pros and Cons Controversy on Molecular Imaging and Dynamic
Open Access Archives of Biotechnology and Biomedicine Research Article Pros and Cons Controversy on Molecular Imaging and Dynamics of Double- ISSN Standard DNA/RNA of Human Preserving 2639-6777 Stem Cells-Binding Nano Molecules with Androgens/Anabolic Steroids (AAS) or Testosterone Derivatives through Tracking of Helium-4 Nucleus (Alpha Particle) Using Synchrotron Radiation Alireza Heidari* Faculty of Chemistry, California South University, 14731 Comet St. Irvine, CA 92604, USA *Address for Correspondence: Dr. Alireza Abstract Heidari, Faculty of Chemistry, California South University, 14731 Comet St. Irvine, CA 92604, In the current study, we have investigated pros and cons controversy on molecular imaging and dynamics USA, Email: of double-standard DNA/RNA of human preserving stem cells-binding Nano molecules with Androgens/ [email protected]; Anabolic Steroids (AAS) or Testosterone derivatives through tracking of Helium-4 nucleus (Alpha particle) using [email protected] synchrotron radiation. In this regard, the enzymatic oxidation of double-standard DNA/RNA of human preserving Submitted: 31 October 2017 stem cells-binding Nano molecules by haem peroxidases (or heme peroxidases) such as Horseradish Peroxidase Approved: 13 November 2017 (HPR), Chloroperoxidase (CPO), Lactoperoxidase (LPO) and Lignin Peroxidase (LiP) is an important process from Published: 15 November 2017 both the synthetic and mechanistic point of view. Copyright: 2017 Heidari A. This is an open access article distributed under the Creative -

Adulteration of Dietary Supplements by the Illegal Addition of Synthetic Drugs: a Review Tiago Rocha, Joana S
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Biblioteca Digital do IPB Adulteration of Dietary Supplements by the Illegal Addition of Synthetic Drugs: A Review Tiago Rocha, Joana S. Amaral, and Maria Beatriz P.P. Oliveira Abstract: In the last few years, the consumption of dietary supplements, especially those having plants as ingredients, has been increasing due to the common idea that they are natural products posing no risks to human health. In the European Union and the United States, dietary supplements are legally considered as foods/special category of foods, thus are not being submitted to any safety assessment prior to their commercialization. Among the issues that can affect safety, adulteration by the illegal addition of pharmaceutical substances or their analogs is of major concern since unscrupulous producers can falsify these products to provide for quick effects and to increase sales. This review discusses the various classes of synthetic drugs most frequently described as being illegally added to dietary supplements marketed for weight loss, muscle building/sport performance and sexual performance enhancement. Information regarding regulation and consumption is also presented. Finally, several conventional and advanced analytical techniques used to detect and identify different adulterants in dietary supplements and therefore also in foods, with particular emphasis on plant food supplements, are critically described. This review demonstrates that dietary supplement adulteration is an emerging food safety problem and that an effective control by food regulatory authorities is needed to safeguard consumers. Keywords: adulteration, analogs, dietary supplements, food safety, pharmaceutical drugs, plant food supplements Introduction and Education Act (DSHEA), respectively, thus not requiring any In the last few years, the consumption of dietary supplements, safety assessment prior to their commercialization. -
C:\Documents and Settings\Ms785a\Desktop\Web
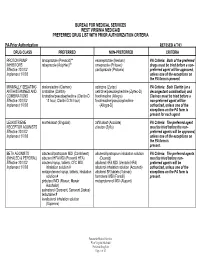
BUREAU FOR MEDICAL SERVICES WEST VIRGINIA MEDICAID PREFERRED DRUG LIST WITH PRIOR AUTHORIZATION CRITERIA PA-Prior Authorization REVISED 4/7/03 DRUG CLASS PREFERRED NON-PREFERRED CRITERIA PROTON PUMP lansoprazole (Prevacid)** esomeprazole (Nexium) PA Criteria: Both of the preferred INHIBITORS rabeprazole (AcipHex)** omeprazole (Prilosec) drugs must be tried before a non- Effective 10/1/02 pantoprazole (Protonix) preferred agent will be approved, Implement 1/7/03 unless one of the exceptions on the PA form is present. MINIMALLY SEDATING desloratadine (Clarinex) cetirizine (Zyrtec) PA Criteria: Both Claritin (or a ANTIHISTAMINES AND loratadine (Claritin) cetirizine/pseudoephedrine (Zyrtec-D) decongestant combination) and COMBINATIONS loratadine/pseudoephedrine (Claritin-D fexofenadine (Allegra) Clarinex must be tried before a Effective 10/1/02 12 hour, Claritin-D 24 hour) fexofenadine/pseudoephedrine non-preferred agent will be Implement 1/7/03 (Allegra-D) authorized, unless one of the exceptions on the PA form is present for each agent. LEUKOTRIENE montelukast (Singulair) zafirlukast (Accolate) PA Criteria: The preferred agent RECEPTOR AGONISTS zileuton (Zyflo) must be tried before the non- Effective 10/1/02 preferred agents will be approved, Implement 1/7/03 unless one of the exceptions on the PA form is present. BETA AGONISTS albuterol/ipratropium MDI (Combivent) albuterol/ipratropium inhalation solution PA Criteria: The preferred agents (INHALED & PERORAL) albuterol HFA MDI (Proventil HFA) (Duoneb) must be tried before non- Effective 10/1/02 albuterol syrup, tablets, CFC MDI, albuterol HFA MDI (Ventolin HFA) preferred agents will be Implement 1/7/03 inhalation solution # albuterol inhalation solution (Accuneb) authorized, unless one of the metaproterenol syrup, tablets, inhalation albuterol SR tablets (Volmax) exceptions on the PA form is solution # formoteral MDI (Foradil) present. -

Screening for Synthetic Steroids in Human Urine by LC-ESI-MS/MS
In: W Schänzer, H Geyer, A Gotzmann, U Mareck (eds.) Recent Advances In Doping Analysis (13). Sport und Buch Strauß - Köln 2005 Mario Thevis, Hans Geyer, Ute Mareck and Wilhelm Schänzer Screening for Synthetic Steroids in Human Urine by LC-ESI-MS/MS Institute of Biochemistry, German Sport University, Cologne, Germany Introduction The problem of unknown synthetic steroids in sports has become evident during the last few years. The anabolic steroid norbolethone (13-ethyl-17-hydroxy-18,19-dinor-17α-pregn-4-en- 3-one) was detected in a doping control urine sample in 2002 [1] although this compound has never been commercially available, and tetrahydrogestrinone (THG, 13-ethyl-17-hydroxy- 18,19-dinor-17α-pregn-4,9,11-trien-3-one) substantiated the fact that designer steroids are abused in the world of sport [2-5] in 2004. In February 2005, it was found that another steroid termed ‘madol’ (17α-methyl-5α-androst-2-ene-17β-ol, also known as desoxy- methyltestosterone (DMT)) [6, 7] may also have been used by athletes. The primary methods of drug screening are based on the mass spectrometric identification of known compounds. Conventional assays will not detect unknown compounds differing by as little as 1 or 2 Daltons from previously identified compounds, especially for MS/MS experiments where defined precursor ions are selected for collision-induced dissociation (CID). In this study a complementary screening procedure is reported allowing the detection of new or unknown steroids with distinct nuclei in human urine. Complementary urinary “steroid profiles” are generated from precursor ion scan experiments using LC-ESI-MS/MS, a strategy also commonly employed also in metabolite identification studies. -

Applications Op Gas Chromatography and Mass Spectrometry to Steroids and Other Biologically Important Materials
APPLICATIONS OP GAS CHROMATOGRAPHY AND MASS SPECTROMETRY TO STEROIDS AND OTHER BIOLOGICALLY IMPORTANT MATERIALS G.M.ANTHONY Ph.D THESIS 1973 ProQuest Number: 11017929 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 11017929 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 C 0 N TENTS PAGE INTRODUCTION 1 PART 1 CATECHOLAMINES AND p> - HYDROXY AMINES 34 PART 2 SOME MODIFIED STEROIDS USED AS DRUGS 50 PART 3 STEROID OLEFINES 70 PART 4 OXYGEN SUBSTITUENTS ON THE STEROID NUCLEUS 111 PART 5 STEROID DRUG METABOLISM STUDY 156 CONCLUSIONS 182 EXPERIMENTAL 188 REFERENCES 198 Applications of One Chromatography .<md Mass Spectrometry to Steroids and Other Biologically Important Materials Summery General Techniques "have been developed for the structural analysis of microgram quantities of son© biological materials using gas chromatography end maos spectrometry# The work has been mostly limited to two typc3 of material (i) p»hydroxy*aminea and related catecholamines (il) natural and gy^thatle steroids The latter type* the study of whlefc ttfaatitatee most of this thesis |i prosents special difficulties due to the many positions on the steroid nucleus which may contain a functional group. -

Gli Steroidi Androgeni 2017
GLI STEROIDI ANDROGENI ANABOLIZZANTI Effetti clinici, Classificazione e Meccanismo d’azione Introduzione. Gli androgeni sono una famiglia di farmaci derivati dal testosterone ed esercitano i loro effetti su molti organi e tessuti del corpo umano come i caratteri sessuali e delle riproduzione, il muscolo, l’osso, la pelle ed i capelli, il fegato, il rene il tessuto emopoietico, il sistema immunitario ed il sistema nervoso centrale e periferico (Mooradian, Morley et al. 1987). L’effetto principale di questi ormoni è la mascolinizzazione, cioè l’accentuazione delle caratteristiche fisiche maschili. Nel feto maschile, gli androgeni stimolano lo sviluppo dell’epididimo, delle vie deferenti, delle vescicole seminali e del dotto eiaculatori (derivati dal dotto Wolffiano) e dai genitali maschile esterni (pene, uretra, scroto) (Wilson, Griffin et al. 1981). Durante la pubertà i testicoli aumentano la produzione di androgeni, in particolare del testosterone che stimola la formazione dei caratteri sessuali primari come l’aumento di volume dei testicoli, dei genitali esterni e dei tessuti accessori della riproduzione come la prostata, le vescicole seminali e bulbo uretrale. Inoltre, gli androgeni stimolano la formazione dei caratteri sessuali secondari che comprendono gli effetti anabolici, come lo sviluppo fisico, la crescita lineare e muscolare, sia quelli androgeni, come l’allargamento della laringe e abbassamento del tono della voce, la crescita del pelo in regione facciale, ascellare e pubica, un aumento delle ghiandole sebacee ad aumento di secrezione del grasso della pelle (formazione di acne) e dei capelli, sul sistema nervoso centrale aumentando la libido e l’aggressività. Gli Androgeni. Gli androgeni nel corpo umano comprendono il testosterone, il diidrotestosterone (DHT), l’androstenedione ed il deidroepiandrosterone (DHEA). -

(12) Patent Application Publication (10) Pub. No.: US 2012/0271275 A1 Biggs Et Al
US 20120271275A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0271275 A1 Biggs et al. (43) Pub. Date: Oct. 25, 2012 (54) MEDICAL DEVICES AND METHODS Publication Classification COMPRISING AN ANABOLICAGENT FOR (51) Int. Cl WOUND HEALING A6II 3/58 (2006.01) A6M 5/42 (2006.01) (75) Inventors: Danielle L. Biggs, Collierville, TN A6IP 7/02 (2006.01) 5147 (US); Jared T. Wilsey, Memphis, (52) U.S. Cl. ......................................... 604/506; 514/176 TN (US) (57) ABSTRACT Improved medical devices and methods are provided com (73) Assignee: WARSAW ORTHOPEDIC, INC., prising an anabolic agent for wound healing. These improved Warsaw, IN (US) medical devices and methods can enhance wound healing in wounds from cuts, abrasions, lesions, burns including Sun burn, Surgical incisions, pressure ulcers, diabetic ulcers, trau (21) Appl. No.: 13/093,479 matic wounds, or other injuries or maladies, which can be chronic or non-chronic in origin. In some embodiments, the medical device comprises a drug depot that releases the ana (22) Filed: Apr. 25, 2011 bolic agent over at least 3 days to enhance wound healing. Patent Application Publication Oct. 25, 2012 US 2012/0271275 A1 o?uaegsås?on,ang?eoongoasoonaoºn6unee?-,punoanseneuerreoov?orozouens 2%3%%*{{{3% (siti) are e assad US 2012/0271275 A1 Oct. 25, 2012 MEDICAL DEVICES AND METHODS tions in the A, B, C and/or D rings have been made to increase COMPRISING AN ANABOLICAGENT FOR binding activity to the Steroid receptor and to increase lipid WOUND HEALING solubility of the anabolic steroids and prolong its activity. For example, alkylation at 17-alpha position with methyl or ethyl BACKGROUND groups create orally active compounds because it slows the 0001 Wounds can occur from various types of cuts, abra degradation of the drug by the liver. -
26414 Rev 08 08 Prf1 Torip
I e I n I s o n r C e e 4 t g s 1 o ‡ r o 0 y t t s 1 l 2 t s s 0 - n e - E e 4 l t O 2 l 0 b d 7 x y a e . 1 i h T R v f t i e e r R e M t s d E n a Esterified Estrogens and Methyltestosterone Tablet s‡ such as beard, pubic, chest, and axillary hair, laryngeal enlargement, vocal cord thickening, alterations INFORMATION FOR THE PATIEN T Rx Only III in body musculature, and fat distribution. Drugs in this class also cause retention of nitrogen, sodium, C potassium, phosphorus, and decreased urinary excretion of calcium. Androgens have been reported to ESTROGENS INCREASE THE RISK WHAT YOU SHOULD KNOW ABOUT ESTROGENS III OF ENDOMETRIAL CANCER increase protein anabolism and decrease protein catabolism. Nitrogen balance is improved only when C Close clinical surveillance of all women taking estrogens is important. Adequate diagnostic measures, there is sufficient intake of calories and protein. Androgens are responsible for the growth spurt of Esterified Estrogens and Methyltestosterone Tablets ‡ including endometrial sampling when indicated, should be undertaken to rule out malignancy in all adolescence and for the eventual termination of linear growth which is brought about by fusion of the cases of undiagnosed persistent or recurring abnormal vaginal bleeding. There is no evidence that the epiphyseal growth centers. In children, exogenous androgens accelerate linear growth rates, but may Read this PATIENT INFORMATION before you start taking Esterified use of “natural” estrogens results in a different endometrial risk profile than synthetic estrogens at cause a disproportionate advancement in bone maturation. -

Androgene-Anabole Steroider (AAS) Og Vold 2
Nasjonalt kunnskapssenter for helsetjenesten Rapport nr. 4/2004 Androgene-anabole steroider (AAS) og vold 2 Forord Justisdepartementet opprettet i 2002 en arbeidsgruppe som skulle se på om det er doku- mentert noen sammenheng mellom misbruk av anabole/androgene steroider (AAS) og risiko for økt aggresjon og voldelig adferd. Senter for medisinsk metodevurdering (SMM) ble anmodet om å assistere gruppen. Gruppen besto av følgende personer: Professor Egil Haug, Hormonlaboratoriet, Aker sykehus Professor Jørg Mørland, Div. for rettstoks. og rusmiddelforskning, Folkehelse- instituttet Professor Bjørnar Olaisen (leder), Formann i Rettsmedisinsk kommisjon Seniorforsker cand. med. Kurt I. Myhre, Senter for medisinsk metodevurdering var sekretær for gruppen. Arbeidsgruppen gjennomførte dette arbeidet i løpet av sommeren og høsten 2003, og uttalelsen ble overlevert Justisdepartementet i januar 2004. Foreliggende SMM-rapport er en noe omarbeidet versjon av høringsuttalelsen. Arbeidet ble finansiert av Justisdepartementet. Berit Mørland Direktør Kurt I. Myhre Prosjektsekretær Rapport 4/2004: Androgene-anabole steroider (AAS) og vold 3 Innhold FORORD............................................................................................................................. 2 INNHOLD ........................................................................................................................... 3 SMMS KOMMENTAR ........................................................................................................ 5 Formål ........................................................................................................................ -

Anabolics 2010
William Llewellyn’s ANABOLICS,10th ed. Softcover ISBN-13: 978-0-9828280-0-7 ISBN-10: 0-9828280-0-4 Hardcover ISBN-13: 978-0-9828280-1-4 ISBN-10: 0-9828280-1-2 WILLIAM LLEWELLYN’S ANABOLICS n o i t i d 10th E DISCLAIMER: This information was gathered from sources including, but not limited to medical journals, pharmaceutical reports, laboratory reports, textbooks, as well as interviews with medical experts, athletes, and steroid distributors. Neither the author nor publisher assumes any liability for the information presented. This book is intended to provide a compendium of information for the reader. None of the information is meant to be applied and is for entertainment purposes only. It is not intended to provide nor replace medical advice. Readers are advised that the substances described in this reference book are to be used only under a physician’s care and may be prohibited in certain jurisdictions.Readers should consult with appropriate medical authorities before using any drug, and proper legal authorities on the status of substances described herein. Neither the publisher nor author advocate readers engage in any illegal activities. Copyright © 2011 and published by Molecular Nutrition, LLC in Jupiter, FL 33458. All rights reserved. None of the content in this publication may be reproduced, stored in a retrieval system, resold, redistributed, or transmitted in any form or by any means (digital, electronic, mechanical, photocopy, recorded, or otherwise) without the prior written permission of the publisher. William Llewellyn’s ANABOLICS are trademarks used herein under license. Molecular Nutrition LLC, 5500 Military Trail, Ste. -

(12) United States Patent (10) Patent No.: US 9,592.243 B2 Wilsey (45) Date of Patent: *Mar
USO09592243B2 (12) United States Patent (10) Patent No.: US 9,592.243 B2 Wilsey (45) Date of Patent: *Mar. 14, 2017 (54) MEDICAL DEVICES AND METHODS (58) Field of Classification Search COMPRISING AN ANABOLICAGENT FOR CPC ... A61L 2400/06; A61L 31/06; A61L 31/148: TREATMENT OF AN INURY A61L 31/16: A61L 26/0019; (Continued) (71) Applicant: Warsaw Orthopedic, Inc., Warsaw, IN (US) (56) References Cited (72) Inventor: Jared T. Wilsey, Memphis, TN (US) U.S. PATENT DOCUMENTS 4,624,255 A 11/1986 Schenck et al. (73) Assignee: Warsaw Orthopedic, Inc., Warsaw, IN 4,742,054 A 5, 1988 Naftchi (US) (Continued) (*) Notice: Subject to any disclaimer, the term of this patent is extended or adjusted under 35 FOREIGN PATENT DOCUMENTS U.S.C. 154(b) by 0 days. WO O3005961 A2 1, 2003 WO 2005034998 A2 4/2005 This patent is Subject to a terminal dis WO 2007OO5177 A1 1, 2007 claimer. (21) Appl. No.: 14/085,444 OTHER PUBLICATIONS Chhipa et al “Formulation Optimization of Sustained Release (22) Filed: Nov. 20, 2013 Pellets of Itopride Hydrochloride using Different Polymers,” Jour (65) Prior Publication Data nal of Pharmacy Research 2009 2(8) 1404-1408).* (Continued) US 2014/0107088 A1 Apr. 17, 2014 Primary Examiner — Suzanne Ziska Related U.S. Application Data (63) Continuation-in-part of application No. 13/093,479, (57) ABSTRACT filed on Apr. 25, 2011. Improved medical devices and methods are provided com prising an anabolic agent for healing an injury. These (51) Int. Cl. improved medical devices and methods can enhance healing A6 IK3I/58 (2006.01) in injuries from traumatic soft tissue injury, Surgical injuries, A6 IK 47/34 (2006.01) burn, traumatic brain injuries, musculotendinous injuries, musculoskeletal conditions, bone injury or other injuries or (Continued) maladies, which can be chronic or non-chronic in origin.