BLOC-2 Subunit HPS6 Deficiency Affects The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Molecular Diagnostic Requisition
BAYLOR MIRACA GENETICS LABORATORIES SHIP TO: Baylor Miraca Genetics Laboratories 2450 Holcombe, Grand Blvd. -Receiving Dock PHONE: 800-411-GENE | FAX: 713-798-2787 | www.bmgl.com Houston, TX 77021-2024 Phone: 713-798-6555 MOLECULAR DIAGNOSTIC REQUISITION PATIENT INFORMATION SAMPLE INFORMATION NAME: DATE OF COLLECTION: / / LAST NAME FIRST NAME MI MM DD YY HOSPITAL#: ACCESSION#: DATE OF BIRTH: / / GENDER (Please select one): FEMALE MALE MM DD YY SAMPLE TYPE (Please select one): ETHNIC BACKGROUND (Select all that apply): UNKNOWN BLOOD AFRICAN AMERICAN CORD BLOOD ASIAN SKELETAL MUSCLE ASHKENAZIC JEWISH MUSCLE EUROPEAN CAUCASIAN -OR- DNA (Specify Source): HISPANIC NATIVE AMERICAN INDIAN PLACE PATIENT STICKER HERE OTHER JEWISH OTHER (Specify): OTHER (Please specify): REPORTING INFORMATION ADDITIONAL PROFESSIONAL REPORT RECIPIENTS PHYSICIAN: NAME: INSTITUTION: PHONE: FAX: PHONE: FAX: NAME: EMAIL (INTERNATIONAL CLIENT REQUIREMENT): PHONE: FAX: INDICATION FOR STUDY SYMPTOMATIC (Summarize below.): *FAMILIAL MUTATION/VARIANT ANALYSIS: COMPLETE ALL FIELDS BELOW AND ATTACH THE PROBAND'S REPORT. GENE NAME: ASYMPTOMATIC/POSITIVE FAMILY HISTORY: (ATTACH FAMILY HISTORY) MUTATION/UNCLASSIFIED VARIANT: RELATIONSHIP TO PROBAND: THIS INDIVIDUAL IS CURRENTLY: SYMPTOMATIC ASYMPTOMATIC *If family mutation is known, complete the FAMILIAL MUTATION/ VARIANT ANALYSIS section. NAME OF PROBAND: ASYMPTOMATIC/POPULATION SCREENING RELATIONSHIP TO PROBAND: OTHER (Specify clinical findings below): BMGL LAB#: A COPY OF ORIGINAL RESULTS ATTACHED IF PROBAND TESTING WAS PERFORMED AT ANOTHER LAB, CALL TO DISCUSS PRIOR TO SENDING SAMPLE. A POSITIVE CONTROL MAY BE REQUIRED IN SOME CASES. REQUIRED: NEW YORK STATE PHYSICIAN SIGNATURE OF CONSENT I certify that the patient specified above and/or their legal guardian has been informed of the benefits, risks, and limitations of the laboratory test(s) requested. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

G060. Phenotypic and Genotypic Study of Patients with Hermansky-Pudlak Syndrome J.A. Majerus, Mayo Clinic, Rochester, MN. Introd
G060. Phenotypic and Genotypic Study of Patients with Hermansky-Pudlak Syndrome J.A. Majerus, Mayo Clinic, Rochester, MN. Introduction: Hermansky-Pudlak syndrome (HPS) represents a group of autosomal recessive disorders due to mutations in genes involved in intracellular vesicular trafficking. Clinical presentations include oculocutaneous albinism and bleeding diathesis. There are 10 genetic subtypes of HPS: type 1 (due to mutations in HPS1), type 2 (AP3B1), type 3 (HPS3), type 4 (HPS4), type 5 (HPS5), type 6 (HPS6), type 7 (DTNBP1), type 8 (BLOC1S3), type 9 (BLOC1S6), and type 10 (AP3D1). Eventually all patients with HPS1, 2 and 4 develop pulmonary fibrosis and may require lung transplant. Since severe platelet dense granule deficiency is considered a characteristic feature of of HPS, platelet whole mount transmission electron microscopy (PTEM) has been considered a good initial screening test. Potential HPS positive cases detected by PTEM still need to be confirmed and further classified by genetic testing. The goal of this study is to assess the HPS gene mutation status of the PTEM identified HPS cases in our institution. Methods: Nine patients with PTEM documented severe dense granule deficiencies and one patient with ocular albinism but normal dense granules by PTEM were included in this study. Next Generation Sequencing (NGS) was performed using a targeted panel (Agilent Technologies) encompassing 9 of the HPS genes (HPS1, AP3B1, HPS3, HPS4, HPS5, HPS6, DTNBP1, BLOC1S3, and BLOC1S6). DNA library preparation was performed using the SureSelectXT Target Enrichment System for Illumina Paired-End Multiplexed Sequencing Library (Agilent Technologies). The enriched indexed DNA sample was then sequenced on an Illumina MiSeq or HiSeq 2500 platform. -

Slc15a4, AP-3, and Hermansky-Pudlak Syndrome Proteins Are Required for Toll-Like Receptor Signaling in Plasmacytoid Dendritic Cells
Slc15a4, AP-3, and Hermansky-Pudlak syndrome proteins are required for Toll-like receptor signaling in plasmacytoid dendritic cells Amanda L. Blasius, Carrie N. Arnold, Philippe Georgel1, Sophie Rutschmann2, Yu Xia, Pei Lin, Charles Ross, Xiaohong Li, Nora G. Smart, and Bruce Beutler3 Department of Genetics, The Scripps Research Institute, La Jolla, CA 92037 Contributed by Bruce Beutler, September 17, 2010 (sent for review September 16, 2010) Despite their low frequency, plasmacytoid dendritic cells (pDCs) abrogate TLR7 and TLR9 signaling in pDCs. We show that lyso- produce most of the type I IFN that is detectable in the blood some-related organelle (LRO) trafficking and biogenesis proteins, following viral infection. The endosomal Toll-like receptors (TLRs) such as adapter-related protein complex-3 (AP-3) and Hermansky- TLR7 and TLR9 are required for pDCs, as well as other cell types, to Pudlack syndrome (HPS) proteins of the biogenesis of lysosome- sense viral nucleic acids, but the mechanism by which signaling related organelle complex (BLOC)-1 and BLOC-2 groups, are through these shared receptors results in the prodigious production specifically required for type I IFN and cytokine production in of type I IFN by pDCs is not understood. We designed a genetic pDCs. Moreover, Slc15a4, an obscure solute channel protein, is screen to identify proteins required for the development and essential for TLR-mediated signaling in pDCs. Our data reveal a specialized function of pDCs. One phenovariant, which we named specialized membrane trafficking mechanism necessary for TLR feeble, showed abrogation of both TLR-induced type I IFN and pro- signaling in pDCs, which could explain their unique responses to inflammatory cytokine production by pDCs, while leaving TLR re- viral infection. -

The Life Cycle of Platelet Granules
The life cycle of platelet granules The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Sharda, Anish, and Robert Flaumenhaft. 2018. “The life cycle of platelet granules.” F1000Research 7 (1): 236. doi:10.12688/f1000research.13283.1. http://dx.doi.org/10.12688/ f1000research.13283.1. Published Version doi:10.12688/f1000research.13283.1 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:35981871 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA F1000Research 2018, 7(F1000 Faculty Rev):236 Last updated: 28 FEB 2018 REVIEW The life cycle of platelet granules [version 1; referees: 2 approved] Anish Sharda, Robert Flaumenhaft Division of Hemostasis and Thrombosis, Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, USA First published: 28 Feb 2018, 7(F1000 Faculty Rev):236 (doi: Open Peer Review v1 10.12688/f1000research.13283.1) Latest published: 28 Feb 2018, 7(F1000 Faculty Rev):236 (doi: 10.12688/f1000research.13283.1) Referee Status: Abstract Invited Referees Platelet granules are unique among secretory vesicles in both their content and 1 2 their life cycle. Platelets contain three major granule types—dense granules, α-granules, and lysosomes—although other granule types have been reported. version 1 Dense granules and α-granules are the most well-studied and the most published physiologically important. -

1 SUPPLEMENTAL DATA Figure S1. Poly I:C Induces IFN-Β Expression
SUPPLEMENTAL DATA Figure S1. Poly I:C induces IFN-β expression and signaling. Fibroblasts were incubated in media with or without Poly I:C for 24 h. RNA was isolated and processed for microarray analysis. Genes showing >2-fold up- or down-regulation compared to control fibroblasts were analyzed using Ingenuity Pathway Analysis Software (Red color, up-regulation; Green color, down-regulation). The transcripts with known gene identifiers (HUGO gene symbols) were entered into the Ingenuity Pathways Knowledge Base IPA 4.0. Each gene identifier mapped in the Ingenuity Pathways Knowledge Base was termed as a focus gene, which was overlaid into a global molecular network established from the information in the Ingenuity Pathways Knowledge Base. Each network contained a maximum of 35 focus genes. 1 Figure S2. The overlap of genes regulated by Poly I:C and by IFN. Bioinformatics analysis was conducted to generate a list of 2003 genes showing >2 fold up or down- regulation in fibroblasts treated with Poly I:C for 24 h. The overlap of this gene set with the 117 skin gene IFN Core Signature comprised of datasets of skin cells stimulated by IFN (Wong et al, 2012) was generated using Microsoft Excel. 2 Symbol Description polyIC 24h IFN 24h CXCL10 chemokine (C-X-C motif) ligand 10 129 7.14 CCL5 chemokine (C-C motif) ligand 5 118 1.12 CCL5 chemokine (C-C motif) ligand 5 115 1.01 OASL 2'-5'-oligoadenylate synthetase-like 83.3 9.52 CCL8 chemokine (C-C motif) ligand 8 78.5 3.25 IDO1 indoleamine 2,3-dioxygenase 1 76.3 3.5 IFI27 interferon, alpha-inducible -

Hermansky-Pudlak Syndrome
Chapter 8 Hermansky-Pudlak Syndrome Naoki Oiso and Akira Kawada Additional information is available at the end of the chapter http://dx.doi.org/10.5772/53573 1. Introduction Oculocutaneous albinism is classified into non-syndromic oculocutaneous albinism (OCA) and syndromic OCA including Hermansky-Pudlak syndrome (HPS), Chediak-Higashi syndrome (CHS) and Griscelli syndrome (GS). Both non-syndromic and syndromic OCAs are autosomal recessive disorders. Human HPS is genetically divided into nine forms, HPS type 1 (HPS-1) to HPS-9. Human HPS can be sub-classified into four subgroups which are associated with protein complexes encoded by the causative genes. In this session, we summarize (1) the clinical features of HPS, (2) the mice and rat models of HPS, and (3) the molecular functions. 2. The clinical features of HPS In 1959, Hermansky and Pudklak described two cases of OCA associated with hemorrhagic diathesis.1 Currently, the condition is known as HPS. HPS is a rare heterogeneous autosomal recessive syndrome which is typically characterized by OCA, bleeding diathesis, and lysosomal ceroid storage resulting from defects of multiple cytoplasmic organelles: melanosomes, platelet dense core granules, and lysosomes.2 The storage of ceroid-like material in lysosomes induces restrictive lung disease, ulcerative colitis, kidney failure, and cardiomyopathy. Accumulation of mice models, identification of causative genes and functional analysis indicated that HPS could be sub-classified into four groups according to four protein complexes, biogenesis of lysosome-related organelles complex-3 (BLOC-3) (HPS-1 and HPS- 4), adaptor protein-3 (AP-3) (HPS-2), BLOC-2 (HPS-3, HPS-5 and HPS-6) and BLOC-1( HPS- 7, HPS-8 and HPS-9).3-5 Currently, more than 16 mice strains and more than 2 rat strains are known as models of human HPS (Table 1). -
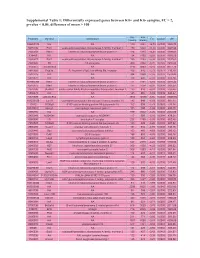
Differentially Expressed Genes Between Kit+ and Kit- Samples, FC
Supplemental Table 1: Differentially expressed genes between Kit+ and Kit- samples, FC > 2, p-value < 0.05, difference of mean > 100 Kit- Kit+ Probeset Symbol Genename FC pvalue diff (mean) (mean) 104280139 NA NA 130 1651 -12,70 0,0000 1520,78 5670239 Ear2 eosinophil-associated, ribonuclease A family, member 2 150 1848 -12,28 0,0000 1697,94 2340358 Ifitm3 interferon induced transmembrane protein 3 126 1315 -10,41 0,0000 1189,02 130465 NA NA 134 1155 -8,60 0,0000 1020,25 2360471 Ear1 eosinophil-associated, ribonuclease A family, member 1 155 1293 -8,34 0,0000 1137,61 7040095 Kit kit oncogene 409 3364 -8,22 0,0145 2955,05 1230347 LOC545854 NA 1216 9893 -8,14 0,0000 8677,76 2810059 Fcgr3a Fc fragment of IgG, low affinity IIIa, receptor 106 816 -7,73 0,0077 710,20 2510725 NA NA 394 2969 -7,53 0,0037 2574,43 5420372 NA NA 128 942 -7,33 0,0000 813,19 101990390 Ifitm2 interferon induced transmembrane protein 2 171 1181 -6,93 0,0000 1010,43 6510075 Ifitm1 interferon induced transmembrane protein 1 152 1054 -6,92 0,0004 901,68 2370286 Slc40a1 solute carrier family 40 (iron-regulated transporter), member 1 133 916 -6,91 0,0000 783,46 5860673 NA NA 143 985 -6,89 0,0239 842,27 6370309 LOC545854 NA 1850 12680 -6,85 0,0000 10829,66 101230129 Ear10 eosinophil-associated, ribonuclease A family, member 10 142 949 -6,68 0,0000 807,11 70112 S100a8 S100 calcium binding protein A8 (calgranulin A) 162 938 -5,79 0,0063 776,16 103780671 Mpeg1 macrophage expressed gene 1 105 598 -5,69 0,0000 493,10 1690184 NA NA 579 3042 -5,26 0,0002 2463,90 2450148 AI324046 expressed -

Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors
University of Cincinnati Date: 12/20/2010 I, Arturo R Maldonado , hereby submit this original work as part of the requirements for the degree of Doctor of Philosophy in Developmental Biology. It is entitled: Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors Student's name: Arturo R Maldonado This work and its defense approved by: Committee chair: Jeffrey Whitsett Committee member: Timothy Crombleholme, MD Committee member: Dan Wiginton, PhD Committee member: Rhonda Cardin, PhD Committee member: Tim Cripe 1297 Last Printed:1/11/2011 Document Of Defense Form Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors A dissertation submitted to the Graduate School of the University of Cincinnati College of Medicine in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (PH.D.) in the Division of Molecular & Developmental Biology 2010 By Arturo Rafael Maldonado B.A., University of Miami, Coral Gables, Florida June 1993 M.D., New Jersey Medical School, Newark, New Jersey June 1999 Committee Chair: Jeffrey A. Whitsett, M.D. Advisor: Timothy M. Crombleholme, M.D. Timothy P. Cripe, M.D. Ph.D. Dan Wiginton, Ph.D. Rhonda D. Cardin, Ph.D. ABSTRACT Since 1999, cancer has surpassed heart disease as the number one cause of death in the US for people under the age of 85. Malignant Peripheral Nerve Sheath Tumor (MPNST), a common malignancy in patients with Neurofibromatosis, and colorectal cancer are midkine- producing tumors with high mortality rates. In vitro and preclinical xenograft models of MPNST were utilized in this dissertation to study the role of midkine (MDK), a tumor-specific gene over- expressed in these tumors and to test the efficacy of a MDK-transcriptionally targeted oncolytic HSV-1 (oHSV). -
Lecture6-For Web.Key
1 2 Holly Wichman Jim Bull 3 4 Phage Population Growth at 43° C A A* H B K ! X174 C G D E J F 5 6 7 8 9 10 Adaptation for cryptic coloration 11 12 Mouse coat color genetics Endothelin receptor B (piebald) mutation Genes Phenotypes Mouse Adamts20, Ectodysplasin-A (Eda), Endothelin 3 Human ligand (Edn3),Endothelin receptor B (Ednrb) Epidermal growth factor receptor (Egfr), Fibroblast growth factor receptor2 (Fgfr2), Inhibitor of kappaB kinase gamma (Ikbkg), C-kit receptor (kit), Ligand for c-kit receptor (kitl), Keratin complex 2, gene 17 (Krt2-17), LIM homeodomain protein 1 (Lmx1a), Mucolipin 3(Mcoln3), Pax-3 transcription factor (Pax3), Sideroflexin (Sfxn1), Neural crest transcription factor (Snai2), Sry-box containing gene 10 (Sox10), Sry-box containing gene 18 (Sox18), T- box 15 (tbx15), Growth factor (Wnt1), Growth factor (Wnt3a), Tyrosinase-related protein 2 (Tyrp2/Dct), Glycoprotein (Gpnmb), Membrane- assoc. transporter protein (Matp), Member of RAS oncogene family (Rab38), Silver protein (Pmel17), Tyrosinase (Tyr), Tyrosinas-related protein 1 (Tyrp1), Beta 3 subunit of adaptor protein 3 (Ap3b1), Delta subunit of adaptor protein 2 (Ap3d), Vacuoloar protein sorting 33a (Vps33a), Cno, Hermansky-Pudlak syndrome gene 1 (Hps1), Hermansky-Pudlak gene 3 (Hps3), Hermansky-Pudlak gene 4 (Hps4), Hermansky- Pudlak gene 5 (Hps5), Hermansky-Pudlak gene 6 (Hps6), Lysosomal trafficking regulator (Lyst), Ocular albinism type 1 (Oa1), Pallidin (Pldn), Rab geranylgeranyl transferase (Rabgtta), Melanophilin (Mlph), Myosin type Va (Myo5a), Myosin type