Ipratropium Bromide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(CD-P-PH/PHO) Report Classification/Justifica
COMMITTEE OF EXPERTS ON THE CLASSIFICATION OF MEDICINES AS REGARDS THEIR SUPPLY (CD-P-PH/PHO) Report classification/justification of medicines belonging to the ATC group R01 (Nasal preparations) Table of Contents Page INTRODUCTION 5 DISCLAIMER 7 GLOSSARY OF TERMS USED IN THIS DOCUMENT 8 ACTIVE SUBSTANCES Cyclopentamine (ATC: R01AA02) 10 Ephedrine (ATC: R01AA03) 11 Phenylephrine (ATC: R01AA04) 14 Oxymetazoline (ATC: R01AA05) 16 Tetryzoline (ATC: R01AA06) 19 Xylometazoline (ATC: R01AA07) 20 Naphazoline (ATC: R01AA08) 23 Tramazoline (ATC: R01AA09) 26 Metizoline (ATC: R01AA10) 29 Tuaminoheptane (ATC: R01AA11) 30 Fenoxazoline (ATC: R01AA12) 31 Tymazoline (ATC: R01AA13) 32 Epinephrine (ATC: R01AA14) 33 Indanazoline (ATC: R01AA15) 34 Phenylephrine (ATC: R01AB01) 35 Naphazoline (ATC: R01AB02) 37 Tetryzoline (ATC: R01AB03) 39 Ephedrine (ATC: R01AB05) 40 Xylometazoline (ATC: R01AB06) 41 Oxymetazoline (ATC: R01AB07) 45 Tuaminoheptane (ATC: R01AB08) 46 Cromoglicic Acid (ATC: R01AC01) 49 2 Levocabastine (ATC: R01AC02) 51 Azelastine (ATC: R01AC03) 53 Antazoline (ATC: R01AC04) 56 Spaglumic Acid (ATC: R01AC05) 57 Thonzylamine (ATC: R01AC06) 58 Nedocromil (ATC: R01AC07) 59 Olopatadine (ATC: R01AC08) 60 Cromoglicic Acid, Combinations (ATC: R01AC51) 61 Beclometasone (ATC: R01AD01) 62 Prednisolone (ATC: R01AD02) 66 Dexamethasone (ATC: R01AD03) 67 Flunisolide (ATC: R01AD04) 68 Budesonide (ATC: R01AD05) 69 Betamethasone (ATC: R01AD06) 72 Tixocortol (ATC: R01AD07) 73 Fluticasone (ATC: R01AD08) 74 Mometasone (ATC: R01AD09) 78 Triamcinolone (ATC: R01AD11) 82 -

Intranasal Rhinitis Agents
Intranasal Rhinitis Agents Therapeutic Class Review (TCR) February 1, 2020 No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, digital scanning, or via any information storage or retrieval system without the express written consent of Magellan Rx Management. All requests for permission should be mailed to: Magellan Rx Management Attention: Legal Department 6950 Columbia Gateway Drive Columbia, Maryland 21046 The materials contained herein represent the opinions of the collective authors and editors and should not be construed to be the official representation of any professional organization or group, any state Pharmacy and Therapeutics committee, any state Medicaid Agency, or any other clinical committee. This material is not intended to be relied upon as medical advice for specific medical cases and nothing contained herein should be relied upon by any patient, medical professional or layperson seeking information about a specific course of treatment for a specific medical condition. All readers of this material are responsible for independently obtaining medical advice and guidance from their own physician and/or other medical professional in regard to the best course of treatment for their specific medical condition. This publication, inclusive of all forms contained herein, is intended to be educational in nature and is intended to be used for informational purposes only. Send comments and suggestions to [email protected]. -

Early Protective Effects of Tiotropium Bromide in Patients with Airways Hyperres P O N S I V E N E S S
European Review for Medical and Pharmacological Sciences 2004; 8: 259-264 Early protective effects of tiotropium bromide in patients with airways hyperres p o n s i v e n e s s C. TERZANO, A. PETROIANNI, A. RICCI, L. D’ANTONI, L. ALLEGRA* Department of Cardiovascular and Respiratory Sciences, Respiratory Diseases Unit, Fondazione E. Lorillard Spencer Cenci, “La Sapienza” University - Rome (Italy) *Institute of Respiratory Diseases, University of Milan, Ospedale Maggiore IRCCS – Milan (Italy) Abstract. – Tiotropium is an anticholiner- Introduction gic drug for Ch ronic Obstructive Pulmonary Di sease (COPD) patients, with a peak b ron- Ti o t rop ium is a qu atern a ry amm o niu m chodilator effect observed after 1.5 to 2 hours and a long duration of action. The aim of our co ngener of atropine. It has been developed study was to quantify the early protection of a as a lon g-acting an ticho linergic bro n c h o d i l a- single dose of inhaled tiotropium against metha- tor for inhalation by patients with chronic ob- choline-induced bronchoconstriction in asthmat- s t ructive pu lmon ary d isease (COPD). Dru g ic patients with airway hyperresponsiveness. p ro p e rties o f ant icho lin ergic agent s have Ten subjects (7M, 3F), with history of asthma been well-kno wn to man y cultures for many and a baseline FEV (Forced Expiratory Volume 1 1 1 c e n t u r i e s . In the 1920s the d iscovery o f the sec) >80% of pred icted, were enrolled in the s t u d y. -

Effect of Ipratropium Bromide on Mucociliary Clearance and Pulmonary Function in Reversible Airways Obstruction
Thorax: first published as 10.1136/thx.34.4.501 on 1 August 1979. Downloaded from Thorax, 1979, 34, 501-507 Effect of ipratropium bromide on mucociliary clearance and pulmonary function in reversible airways obstruction DEMETRI PAVIA, J RODERICK M BATEMAN, NOIRIN F SHEAHAN, AND STEWART W CLARKE From the Department of Thoracic Medicine, The Royal Free Hospital, London NW3 2QG, UK ABSTRACT The effects of (a) regular use for one week and (b) a single dose of a synthetic anticholinergic (ipratropium bromide) on lung mucociliary clearance and as a bronchodilator was ascertained in a controlled, double-blind, cross-over study in 12 patients with reversible airways obstruction (mean increase in FEV1 after isoprenaline: 17%, range 10-50%). Two puffs from a metered dose inhaler of either placebo (propellants only) or drug (40 ,ug) were administered four times a day for one week (regular use), and mucociliary clearance was measured, by radioaerosol tracer, at the end of each treatment period and after a control period in which no treatment was given. On the mornings of the measurements after the placebo and drug periods one final dose (single dose) of ipratropium (40 ,ug) or placebo was given 2 5 hours before the start of the test. There was no statistically significant difference between the three mean mucociliary clearance curves (control, placebo, and drug) for the group; however, there was a significantly greater penetration towards the periphery of the lung of the tracer in the test was after drug administration compared with the other two. This increased penetration http://thorax.bmj.com/ attributed to bronchodilatation caused by the drug. -

NINDS Custom Collection II
ACACETIN ACEBUTOLOL HYDROCHLORIDE ACECLIDINE HYDROCHLORIDE ACEMETACIN ACETAMINOPHEN ACETAMINOSALOL ACETANILIDE ACETARSOL ACETAZOLAMIDE ACETOHYDROXAMIC ACID ACETRIAZOIC ACID ACETYL TYROSINE ETHYL ESTER ACETYLCARNITINE ACETYLCHOLINE ACETYLCYSTEINE ACETYLGLUCOSAMINE ACETYLGLUTAMIC ACID ACETYL-L-LEUCINE ACETYLPHENYLALANINE ACETYLSEROTONIN ACETYLTRYPTOPHAN ACEXAMIC ACID ACIVICIN ACLACINOMYCIN A1 ACONITINE ACRIFLAVINIUM HYDROCHLORIDE ACRISORCIN ACTINONIN ACYCLOVIR ADENOSINE PHOSPHATE ADENOSINE ADRENALINE BITARTRATE AESCULIN AJMALINE AKLAVINE HYDROCHLORIDE ALANYL-dl-LEUCINE ALANYL-dl-PHENYLALANINE ALAPROCLATE ALBENDAZOLE ALBUTEROL ALEXIDINE HYDROCHLORIDE ALLANTOIN ALLOPURINOL ALMOTRIPTAN ALOIN ALPRENOLOL ALTRETAMINE ALVERINE CITRATE AMANTADINE HYDROCHLORIDE AMBROXOL HYDROCHLORIDE AMCINONIDE AMIKACIN SULFATE AMILORIDE HYDROCHLORIDE 3-AMINOBENZAMIDE gamma-AMINOBUTYRIC ACID AMINOCAPROIC ACID N- (2-AMINOETHYL)-4-CHLOROBENZAMIDE (RO-16-6491) AMINOGLUTETHIMIDE AMINOHIPPURIC ACID AMINOHYDROXYBUTYRIC ACID AMINOLEVULINIC ACID HYDROCHLORIDE AMINOPHENAZONE 3-AMINOPROPANESULPHONIC ACID AMINOPYRIDINE 9-AMINO-1,2,3,4-TETRAHYDROACRIDINE HYDROCHLORIDE AMINOTHIAZOLE AMIODARONE HYDROCHLORIDE AMIPRILOSE AMITRIPTYLINE HYDROCHLORIDE AMLODIPINE BESYLATE AMODIAQUINE DIHYDROCHLORIDE AMOXEPINE AMOXICILLIN AMPICILLIN SODIUM AMPROLIUM AMRINONE AMYGDALIN ANABASAMINE HYDROCHLORIDE ANABASINE HYDROCHLORIDE ANCITABINE HYDROCHLORIDE ANDROSTERONE SODIUM SULFATE ANIRACETAM ANISINDIONE ANISODAMINE ANISOMYCIN ANTAZOLINE PHOSPHATE ANTHRALIN ANTIMYCIN A (A1 shown) ANTIPYRINE APHYLLIC -

Therapeutic Class Overview Inhaled Anticholinergics
Therapeutic Class Overview Inhaled Anticholinergics Therapeutic Class Overview/Summary: The inhaled anticholinergics are a class of bronchodilators primarily used in the management of chronic obstructive pulmonary disease (COPD), a condition characterized by progressive airflow restrictions that are not fully reversible.1-3 Symptoms associated with COPD typically include dyspnea, cough, sputum production, wheezing and chest tightness. Specifically, inhaled anticholinergics work via the inhibition of acetylcholine at parasympathetic sites in bronchial smooth muscle causing bronchodilation. Meaningful increases in lung function can be achieved with the use of inhaled anticholinergics in patients with COPD.1-3 The available single-entity inhaled anticholinergics include aclidinium (Tudorza® Pressair), glycopyrrolate (Seebri Neohaler®), ipratropium (Atrovent®, Atrovent® HFA), tiotropium (Spiriva®, Spiriva Respimat®) and umeclidinium (Incruse Ellipta®) with the combination products including glycopyrrolate/indacaterol (Utibron Neohaler®), umeclidinium/vilanterol (Anoro Ellipta®), tiotropium/olodaterol (Stiolto Respimat®) and ipratropium/albuterol, formulated as either an inhaler (Combivent Respimat®) or nebulizer solution (DuoNeb).4-15 Ipratropium, a short-acting bronchodilator, has a duration of action of six to eight hours and requires administration four times daily. Aclidinium, glycopyrrolate, tiotropium and umeclidinium are considered long-acting bronchodilators. Aclidinium is dosed twice daily, while glycopyrrolate, tiotropium and umeclidinium -

COPD Agents Review – October 2020 Page 2 | Proprietary Information
COPD Agents Therapeutic Class Review (TCR) October 1, 2020 No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, digital scanning, or via any information storage or retrieval system without the express written consent of Magellan Rx Management. All requests for permission should be mailed to: Magellan Rx Management Attention: Legal Department 6950 Columbia Gateway Drive Columbia, Maryland 21046 The materials contained herein represent the opinions of the collective authors and editors and should not be construed to be the official representation of any professional organization or group, any state Pharmacy and Therapeutics committee, any state Medicaid Agency, or any other clinical committee. This material is not intended to be relied upon as medical advice for specific medical cases and nothing contained herein should be relied upon by any patient, medical professional or layperson seeking information about a specific course of treatment for a specific medical condition. All readers of this material are responsible for independently obtaining medical advice and guidance from their own physician and/or other medical professional in regard to the best course of treatment for their specific medical condition. This publication, inclusive of all forms contained herein, is intended to be educational in nature and is intended to be used for informational purposes only. Send comments and suggestions to [email protected]. October 2020 -

Preferred Drug List Effective May 1, 2017
Preferred drug list Effective May 1, 2017 Table of Contents 1.0 Analgesics ………………………………………………………………………………………... 1 2.0 Anesthetics ……………………………………………………………………………………….. 1 3.0 Antibiotics and Antivirals ………………………………………………………………………... 1 4.0 Antineoplastics/Immunosuppressants …………………………………………………………… 2 5.0 Cardiovascular Agents …………………………………………………………………………… 2 6.0 Central Nervous System Agents …………………………………………………………………. 3 7.0 Dermatologicals ………………………………………………………………………………….. 4 8.0 Eyes, Ears, Nose, Mouth and Throat …………………………………………………………….. 4 9.0 Endocrine Agents ………………………………………………………………………………… 5 10.0 Gastrointestinal Agents ………………………………………………………………………….. 5 11.0 Blood Modifiers, Nutritionals and Electrolytes ………………………………………………….. 6 12.0 OB/GYN …………………………………………………………………………………………. 6 13.0 Respiratory Agents ………………………………………………………………………………. 7 14.0 Skeletal Muscle Relaxants ……………………………………………………………………….. 8 15.0 Urologicals ……………………………………………………………………………………….. 8 16.0 Immunologicals, Vaccines and Biotechnology Drugs …………………………………………… 8 17.0 Smoking Cessation ………………………………………………………………………………. 8 www.anthem.com/inmedicaid Anthem Blue Cross and Blue Shield is the trade name of Anthem Insurance Companies, Inc., independent licensee of the Blue Cross and Blue Shield Association. ANTHEM is a registered trademark of Anthem Insurance Companies, Inc. The Blue Cross and Blue Shield names and symbols are registered marks of the Blue Cross and Blue Shield Association. Express Scripts, Inc. is a separate company that provides pharmacy services and pharmacy benefit management -
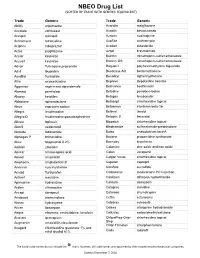
Drug List (SORTED by TRADE with GENERIC EQUIVALENT)
NBEO Drug List (SORTED BY TRADE WITH GENERIC EQUIVALENT) Trade Generic Trade Generic Abilify aripiprazole Avandia rosiglitazone Accolate zafirlukast Avastin bevacizumab Accupril quinapril Azasan azathioprine Achromycin tetracycline AzaSite azithromycin Aciphex rabeprazole Avodart dutasteride Actos pioglitazone Azopt brinzolamide Acular ketorolac Bactrim trimethoprim-sulfamethoxazole Acuvail ketorolac Bactrim DS trimethoprim-sulfamethoxazole Advair fluticasone propionate Baquacil polyhexamethylene biguanide Advil ibuprofen Beconase AQ beclomethasone AeroBid flunisolide Benadryl diphenhydramine Afrin oxymetazoline Bepreve Bepotastine besilate Aggrenox aspirin and dipyridamole Besivance besifloxacin Alamast pemirolast Betadine povidone-iodine Alaway ketotifen Betagan levobunolol Aldactone spironolactone Betasept chlorhexidine topical Aleve naproxen sodium Betaseron interferon beta-1b Allegra fexofenadine Betimol timolol Allegra-D fexofenadine-pseudoephedrine Betoptic S betaxolol Alluvia lopinavir Biopatch chlorhexidine topical Alocril nedocromil Blephamide sulfacetamide-prednisolone Alomide lodoxamide Botox onabotulinum toxinA Alphagan P brimonidine Brolene propamidine isethionate Alrex loteprednol 0.2% Bromday bromfenac Ambien zolpidem Calamine zinc oxide and iron oxide Amicar aminocaproic acid Calan verapamil Amoxil amoxicillin Calgon Vesta chlorhexidine topical Amphocin amphotericin B Capoten captopril Anectine succinylcholine Carafate sucralfate Ansaid flurbiprofen Carbocaine mepivacaine HCl injection Antivert meclizine Cardizem diltiazem -

FDA Briefing Document Pulmonary-Allergy Drugs Advisory Committee Meeting
FDA Briefing Document Pulmonary-Allergy Drugs Advisory Committee Meeting August 31, 2020 sNDA 209482: fluticasone furoate/umeclidinium/vilanterol fixed dose combination to reduce all-cause mortality in patients with chronic obstructive pulmonary disease NDA209482/S-0008 PADAC Clinical and Statistical Briefing Document Fluticasone furoate/umeclidinium/vilanterol fixed dose combination for all-cause mortality DISCLAIMER STATEMENT The attached package contains background information prepared by the Food and Drug Administration (FDA) for the panel members of the advisory committee. The FDA background package often contains assessments and/or conclusions and recommendations written by individual FDA reviewers. Such conclusions and recommendations do not necessarily represent the final position of the individual reviewers, nor do they necessarily represent the final position of the Review Division or Office. We have brought the supplemental New Drug Application (sNDA) 209482, for fluticasone furoate/umeclidinium/vilanterol, as an inhaled fixed dose combination, for the reduction in all-cause mortality in patients with COPD, to this Advisory Committee in order to gain the Committee’s insights and opinions, and the background package may not include all issues relevant to the final regulatory recommendation and instead is intended to focus on issues identified by the Agency for discussion by the advisory committee. The FDA will not issue a final determination on the issues at hand until input from the advisory committee process has been considered -

Application Number
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 203975s000 SUMMARY REVIEW SUMMARY REVIEW OF REGULATORY ACTION Date: November 26, 2013 From: Badrul A. Chowdhury, MD, PhD Director, Division of Pulmonary, Allergy, and Rheumatology Products, CDER, FDA Subject: Division Director Summary Review NDA Number: 20-3975 Applicant Name: GlaxoSmithKline Date of Submission: December 18, 2012 PDUFA Goal Date: December 18, 2013 Proprietary Name: Anoro Ellipta Established Name: Umeclidinium and vilanterol Dosage form: Inhalation Powder (inhaler contains 2 double-foil blister strips, each with 30 blisters containing powder for oral inhalation) Strength: Umeclidinium 62.5 mcg per blister and vilanterol 25 mcg per blister Proposed Indications: Maintenance treatment of airflow obstruction in chronic obstructive pulmonary disease (COPD) Action: Approval 1. Introduction GlaxoSmithKline (GSK) submitted this 505(b)(1) new drug application for use of Anoro Ellipta (umeclidinium 62.5 mcg and vilanterol 25 mcg inhalation powder) for long-term once-daily maintenance bronchodilator treatment of airflow obstruction in patients with chronic obstructive pulmonary disease (COPD). The proposed dose is one inhalation (umeclidinium 62.5 mcg and vilanterol 25 mcg) once daily. The application is based on clinical efficacy and safety studies. This summary review will provide an overview of the application, with a focus on the clinical efficacy and safety studies. 2. Background There are several drug classes available for the relief of airflow obstruction in patients with COPD. These include short- and long-acting beta-2 adrenergic agonists, short- and long-acting anticholinergics, combination products containing beta-2 adrenergic agonists and anticholinergics, combination of long-acting beta-2 adrenergic agonists and corticosteroids, methylxanthines, and phosphodiesterase-4 (PDE4) inhibitors. -

Transient Paradoxical Bronchospasm Associated with Inhalation of The
Jorup et al. BMC Pulmonary Medicine 2014, 14:52 http://www.biomedcentral.com/1471-2466/14/52 RESEARCH ARTICLE Open Access Transient paradoxical bronchospasm associated with inhalation of the LAMA AZD9164: analysis of two Phase I, randomised, double-blind, placebo-controlled studies Carin Jorup1*, Thomas Bengtsson2, Kerstin Strandgården1 and Ulf Sjöbring1 Abstract Background: AZD9164 has demonstrated potential as an inhaled, long-acting, muscarinic antagonist (LAMA) bronchodilator. However, in patients with COPD, but not in healthy subjects, a transient initial drop in FEV1 was observed following inhalation of nebulised doses of AZD9164 in citrate buffer. Two additional studies were conducted to further assess the safety and tolerability of multiple ascending doses of AZD9164 in 27 white and 18 Japanese healthy subjects and in 4 patients with COPD. In these studies, AZD9164 was inhaled via Turbuhaler™. Methods: These were Phase I, randomised, double-blind, placebo-controlled, multiple ascending dose (MAD) studies conducted in Sweden and UK. Healthy subjects (mean age 25.9 yrs) and patients with COPD (mean age 66 yrs, mean post-bronchodilator FEV1 60.1% predicted normal value) were randomised 2:1 to active treatment (400, 1000 or 2800 μg delivered doses of AZD9164) or placebo. Results: No safety or tolerability concerns were identified in the healthy subjects at doses up to and including 2800 μg and both studies confirmed the bronchodilator effect of AZD9164. However, the first 3 patients in the COPD cohort who received AZD9164 (1000 μg) experienced a transient fall in FEV1 5to15minutesafter inhalation of AZD9164 while the patient receiving placebo did not. The study safety review process then resulted in cessation of further activities on AZD9164.