Efficient Adaptive Designs for Clinical Trials of Interventions for COVID-19
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Adaptive Clinical Trials: an Introduction
Adaptive clinical trials: an introduction What are the advantages and disadvantages of adaptive clinical trial designs? How and why were Introduction adaptive clinical Adaptive clinical trial design is trials developed? becoming a hot topic in healthcare research, with some researchers In 2004, the FDA published a report arguing that adaptive trials have the on the problems faced by the potential to get new drugs to market scientific community in developing quicker. In this article, we explain new medical treatments.2 The what adaptive trials are and why they report highlighted that the pace of were developed, and we explore both innovation in biomedical science is the advantages of adaptive designs outstripping the rate of advances and the concerns being raised by in the available technologies and some in the healthcare community. tools for evaluating new treatments. Outdated tools are being used to assess new treatments and there What are adaptive is a critical need to improve the effectiveness and efficiency of clinical trials? clinical trials.2 The current process for developing Adaptive clinical trials enable new treatments is expensive, takes researchers to change an aspect a long time and in some cases, the of a trial design at an interim development process has to be assessment, while controlling stopped after significant amounts the rate of type 1 errors.1 Interim of time and resources have been assessments can help to determine invested.2 In 2006, the FDA published whether a trial design is the most a “Critical Path Opportunities -

FDA Oncology Experience in Innovative Adaptive Trial Designs
Innovative Adaptive Trial Designs Rajeshwari Sridhara, Ph.D. Director, Division of Biometrics V Office of Biostatistics, CDER, FDA 9/3/2015 Sridhara - Ovarian cancer workshop 1 Fixed Sample Designs • Patient population, disease assessments, treatment, sample size, hypothesis to be tested, primary outcome measure - all fixed • No change in the design features during the study Adaptive Designs • A study that includes a prospectively planned opportunity for modification of one or more specified aspects of the study design and hypotheses based on analysis of data (interim data) from subjects in the study 9/3/2015 Sridhara - Ovarian cancer workshop 2 Bayesian Designs • In the Bayesian paradigm, the parameter measuring treatment effect is regarded as a random variable • Bayesian inference is based on the posterior distribution (Bayes’ Rule – updated based on observed data) – Outcome adaptive • By definition adaptive design 9/3/2015 Sridhara - Ovarian cancer workshop 3 Adaptive Designs (Frequentist or Bayesian) • Allows for planned design modifications • Modifications based on data accrued in the trial up to the interim time • Unblinded or blinded interim results • Control probability of false positive rate for multiple options • Control operational bias • Assumes independent increments of information 9/3/2015 Sridhara - Ovarian cancer workshop 4 Enrichment Designs – Prognostic or Predictive • Untargeted or All comers design: – post-hoc enrichment, prospective-retrospective designs – Marker evaluation after randomization (example: KRAS in cetuximab -
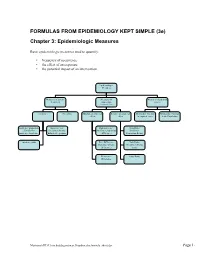
FORMULAS from EPIDEMIOLOGY KEPT SIMPLE (3E) Chapter 3: Epidemiologic Measures
FORMULAS FROM EPIDEMIOLOGY KEPT SIMPLE (3e) Chapter 3: Epidemiologic Measures Basic epidemiologic measures used to quantify: • frequency of occurrence • the effect of an exposure • the potential impact of an intervention. Epidemiologic Measures Measures of disease Measures of Measures of potential frequency association impact (“Measures of Effect”) Incidence Prevalence Absolute measures of Relative measures of Attributable Fraction Attributable Fraction effect effect in exposed cases in the Population Incidence proportion Incidence rate Risk difference Risk Ratio (Cumulative (incidence density, (Incidence proportion (Incidence Incidence, Incidence hazard rate, person- difference) Proportion Ratio) Risk) time rate) Incidence odds Rate Difference Rate Ratio (Incidence density (Incidence density difference) ratio) Prevalence Odds Ratio Difference Macintosh HD:Users:buddygerstman:Dropbox:eks:formula_sheet.doc Page 1 of 7 3.1 Measures of Disease Frequency No. of onsets Incidence Proportion = No. at risk at beginning of follow-up • Also called risk, average risk, and cumulative incidence. • Can be measured in cohorts (closed populations) only. • Requires follow-up of individuals. No. of onsets Incidence Rate = ∑person-time • Also called incidence density and average hazard. • When disease is rare (incidence proportion < 5%), incidence rate ≈ incidence proportion. • In cohorts (closed populations), it is best to sum individual person-time longitudinally. It can also be estimated as Σperson-time ≈ (average population size) × (duration of follow-up). Actuarial adjustments may be needed when the disease outcome is not rare. • In an open populations, Σperson-time ≈ (average population size) × (duration of follow-up). Examples of incidence rates in open populations include: births Crude birth rate (per m) = × m mid-year population size deaths Crude mortality rate (per m) = × m mid-year population size deaths < 1 year of age Infant mortality rate (per m) = × m live births No. -

Incidence and Secondary Transmission of SARS-Cov-2 Infections in Schools
Prepublication Release Incidence and Secondary Transmission of SARS-CoV-2 Infections in Schools Kanecia O. Zimmerman, MD; Ibukunoluwa C. Akinboyo, MD; M. Alan Brookhart, PhD; Angelique E. Boutzoukas, MD; Kathleen McGann, MD; Michael J. Smith, MD, MSCE; Gabriela Maradiaga Panayotti, MD; Sarah C. Armstrong, MD; Helen Bristow, MPH; Donna Parker, MPH; Sabrina Zadrozny, PhD; David J. Weber, MD, MPH; Daniel K. Benjamin, Jr., MD, PhD; for The ABC Science Collaborative DOI: 10.1542/peds.2020-048090 Journal: Pediatrics Article Type: Regular Article Citation: Zimmerman KO, Akinboyo IC, Brookhart A, et al. Incidence and secondary transmission of SARS-CoV-2 infections in schools. Pediatrics. 2021; doi: 10.1542/peds.2020- 048090 This is a prepublication version of an article that has undergone peer review and been accepted for publication but is not the final version of record. This paper may be cited using the DOI and date of access. This paper may contain information that has errors in facts, figures, and statements, and will be corrected in the final published version. The journal is providing an early version of this article to expedite access to this information. The American Academy of Pediatrics, the editors, and authors are not responsible for inaccurate information and data described in this version. Downloaded from©2021 www.aappublications.org/news American Academy by of guest Pediatrics on September 27, 2021 Prepublication Release Incidence and Secondary Transmission of SARS-CoV-2 Infections in Schools Kanecia O. Zimmerman, MD1,2,3; Ibukunoluwa C. Akinboyo, MD1,2; M. Alan Brookhart, PhD4; Angelique E. Boutzoukas, MD1,2; Kathleen McGann, MD2; Michael J. -

Estimated HIV Incidence and Prevalence in the United States
Volume 26, Number 1 Estimated HIV Incidence and Prevalence in the United States, 2015–2019 This issue of the HIV Surveillance Supplemental Report is published by the Division of HIV/AIDS Prevention, National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention, Centers for Disease Control and Prevention (CDC), U.S. Department of Health and Human Services, Atlanta, Georgia. Estimates are presented for the incidence and prevalence of HIV infection among adults and adolescents (aged 13 years and older) based on data reported to CDC through December 2020. The HIV Surveillance Supplemental Report is not copyrighted and may be used and reproduced without permission. Citation of the source is, however, appreciated. Suggested citation Centers for Disease Control and Prevention. Estimated HIV incidence and prevalence in the United States, 2015–2019. HIV Surveillance Supplemental Report 2021;26(No. 1). http://www.cdc.gov/ hiv/library/reports/hiv-surveillance.html. Published May 2021. Accessed [date]. On the Web: http://www.cdc.gov/hiv/library/reports/hiv-surveillance.html Confidential information, referrals, and educational material on HIV infection CDC-INFO 1-800-232-4636 (in English, en Español) 1-888-232-6348 (TTY) http://wwwn.cdc.gov/dcs/ContactUs/Form Acknowledgments Publication of this report was made possible by the contributions of the state and territorial health departments and the HIV surveillance programs that provided surveillance data to CDC. This report was prepared by the following staff and contractors of the Division of HIV/AIDS Prevention, National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention, CDC: Laurie Linley, Anna Satcher Johnson, Ruiguang Song, Sherry Hu, Baohua Wu, H. -

The Statistical Significance of Randomized Controlled Trial Results Is Frequently Fragile: a Case for a Fragility Index
The statistical significance of randomized controlled trial results is frequently fragile: A case for a Fragility Index Walsh, M., Srinathan, S. K., McAuley, D. F., Mrkobrada, M., Levine, O., Ribic, C., Molnar, A. O., Dattani, N. D., Burke, A., Guyatt, G., Thabane, L., Walter, S. D., Pogue, J., & Devereaux, P. J. (2014). The statistical significance of randomized controlled trial results is frequently fragile: A case for a Fragility Index. Journal of Clinical Epidemiology, 67(6), 622-628. https://doi.org/10.1016/j.jclinepi.2013.10.019 Published in: Journal of Clinical Epidemiology Document Version: Publisher's PDF, also known as Version of record Queen's University Belfast - Research Portal: Link to publication record in Queen's University Belfast Research Portal Publisher rights 2014 The Authors. Published by Elsevier Inc. This is an open access article published under a Creative Commons Attribution-NonCommercial-NoDerivs License (https://creativecommons.org/licenses/by-nc-nd/3.0/), which permits distribution and reproduction for non-commercial purposes, provided the author and source are cited. General rights Copyright for the publications made accessible via the Queen's University Belfast Research Portal is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The Research Portal is Queen's institutional repository that provides access to Queen's research output. Every effort has been made to ensure that content in the Research Portal does not infringe any person's rights, or applicable UK laws. -

Evolution of Clinical Trials Throughout History
Evolution of clinical trials throughout history Emma M. Nellhaus1, Todd H. Davies, PhD1 Author Affiliations: 1. Office of Research and Graduate Education, Marshall University Joan C. Edwards School of Medicine, Huntington, West Virginia The authors have no financial disclosures to declare and no conflicts of interest to report. Corresponding Author: Todd H. Davies, PhD Director of Research Development and Translation Marshall University Joan C. Edwards School of Medicine Huntington, West Virginia Email: [email protected] Abstract The history of clinical research accounts for the high ethical, scientific, and regulatory standards represented in current practice. In this review, we aim to describe the advances that grew from failures and provide a comprehensive view of how the current gold standard of clinical practice was born. This discussion of the evolution of clinical trials considers the length of time and efforts that were made in order to designate the primary objective, which is providing improved care for our patients. A gradual, historic progression of scientific methods such as comparison of interventions, randomization, blinding, and placebos in clinical trials demonstrates how these techniques are collectively responsible for a continuous advancement of clinical care. Developments over the years have been ethical as well as clinical. The Belmont Report, which many investigators lack appreciation for due to time constraints, represents the pinnacle of ethical standards and was developed due to significant misconduct. Understanding the history of clinical research may help investigators value the responsibility of conducting human subjects’ research. Keywords Clinical Trials, Clinical Research, History, Belmont Report In modern medicine, the clinical trial is the gold standard and most dominant form of clinical research. -

Quasi-Experimental Studies in the Fields of Infection Control and Antibiotic Resistance, Ten Years Later: a Systematic Review
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Infect Control Manuscript Author Hosp Epidemiol Manuscript Author . Author manuscript; available in PMC 2019 November 12. Published in final edited form as: Infect Control Hosp Epidemiol. 2018 February ; 39(2): 170–176. doi:10.1017/ice.2017.296. Quasi-experimental Studies in the Fields of Infection Control and Antibiotic Resistance, Ten Years Later: A Systematic Review Rotana Alsaggaf, MS, Lyndsay M. O’Hara, PhD, MPH, Kristen A. Stafford, PhD, MPH, Surbhi Leekha, MBBS, MPH, Anthony D. Harris, MD, MPH, CDC Prevention Epicenters Program Department of Epidemiology and Public Health, University of Maryland School of Medicine, Baltimore, Maryland. Abstract OBJECTIVE.—A systematic review of quasi-experimental studies in the field of infectious diseases was published in 2005. The aim of this study was to assess improvements in the design and reporting of quasi-experiments 10 years after the initial review. We also aimed to report the statistical methods used to analyze quasi-experimental data. DESIGN.—Systematic review of articles published from January 1, 2013, to December 31, 2014, in 4 major infectious disease journals. METHODS.—Quasi-experimental studies focused on infection control and antibiotic resistance were identified and classified based on 4 criteria: (1) type of quasi-experimental design used, (2) justification of the use of the design, (3) use of correct nomenclature to describe the design, and (4) statistical methods used. RESULTS.—Of 2,600 articles, 173 (7%) featured a quasi-experimental design, compared to 73 of 2,320 articles (3%) in the previous review (P<.01). Moreover, 21 articles (12%) utilized a study design with a control group; 6 (3.5%) justified the use of a quasi-experimental design; and 68 (39%) identified their design using the correct nomenclature. -

Why Do You Need a Biostatistician? Antonia Zapf1* , Geraldine Rauch2 and Meinhard Kieser3
Zapf et al. BMC Medical Research Methodology (2020) 20:23 https://doi.org/10.1186/s12874-020-0916-4 DEBATE Open Access Why do you need a biostatistician? Antonia Zapf1* , Geraldine Rauch2 and Meinhard Kieser3 Abstract The quality of medical research importantly depends, among other aspects, on a valid statistical planning of the study, analysis of the data, and reporting of the results, which is usually guaranteed by a biostatistician. However, there are several related professions next to the biostatistician, for example epidemiologists, medical informaticians and bioinformaticians. For medical experts, it is often not clear what the differences between these professions are and how the specific role of a biostatistician can be described. For physicians involved in medical research, this is problematic because false expectations often lead to frustration on both sides. Therefore, the aim of this article is to outline the tasks and responsibilities of biostatisticians in clinical trials as well as in other fields of application in medical research. Keywords: Medical research, Biostatistician, Tasks, Responsibilities Background Biometric Society (IBS) provides a definition of biomet- What is a biostatistician, what does he or she actually do rics as a ‘field of development of statistical and mathem- and what distinguishes him or her from, for example, an atical methods applicable in the biological sciences’ [2]. epidemiologist? If we would ask this our main cooper- In here, we will focus on (human) medicine as area of ation partners like physicians or biologists, they probably application, but the results can be easily transferred to could not give a satisfying answer. This is problematic the other biological sciences like, for example, agricul- because false expectations often lead to frustration on ture or ecology. -

A Guide to Systematic Review and Meta-Analysis of Prediction Model Performance
RESEARCH METHODS AND REPORTING A guide to systematic review and meta-analysis of prediction model performance Thomas P A Debray,1,2 Johanna A A G Damen,1,2 Kym I E Snell,3 Joie Ensor,3 Lotty Hooft,1,2 Johannes B Reitsma,1,2 Richard D Riley,3 Karel G M Moons1,2 1Cochrane Netherlands, Validation of prediction models is diagnostic test accuracy studies. Compared to therapeu- University Medical Center tic intervention and diagnostic test accuracy studies, Utrecht, PO Box 85500 Str highly recommended and increasingly there is limited guidance on the conduct of systematic 6.131, 3508 GA Utrecht, Netherlands common in the literature. A systematic reviews and meta-analysis of primary prognosis studies. 2Julius Center for Health review of validation studies is therefore A common aim of primary prognostic studies con- Sciences and Primary Care, cerns the development of so-called prognostic predic- University Medical Center helpful, with meta-analysis needed to tion models or indices. These models estimate the Utrecht, PO Box 85500 Str 6.131, 3508 GA Utrecht, summarise the predictive performance individualised probability or risk that a certain condi- Netherlands of the model being validated across tion will occur in the future by combining information 3Research Institute for Primary from multiple prognostic factors from an individual. Care and Health Sciences, Keele different settings and populations. This Unfortunately, there is often conflicting evidence about University, Staffordshire, UK article provides guidance for the predictive performance of developed prognostic Correspondence to: T P A Debray [email protected] researchers systematically reviewing prediction models. For this reason, there is a growing Additional material is published demand for evidence synthesis of (external validation) online only. -

U.S. Investments in Medical and Health Research and Development 2013 - 2017 Advocacy Has Helped Bring About Five Years of Much-Needed Growth in U.S
Fall 2018 U.S. Investments in Medical and Health Research and Development 2013 - 2017 Advocacy has helped bring about five years of much-needed growth in U.S. medical and health research investment. More advocacy is critical now to ensure our nation steps up in response to health threats that we can—we must—overcome. More Than Half Favor Doubling Federal Spending on Medical Research Do you favor or oppose doubling federal spending on medical research over the next five years? 19% Not Sure 23% 8% Strongly favor Strongly oppose 16% 35% Somewhat oppose Somewhat favor Source: A Research!America survey of U.S. adults conducted in partnership with Zogby Analytics in January 2018. Research!America 3 Introduction Investment1 in medical and health research and development (R&D) in the U.S. grew by $38.8 billion or 27% from 2013 to 2017. Industry continues to invest more than any other sector, accounting for 67% of total spending in 2017, followed by the federal government at 22%. Federal investments increased from 2016 to 2017, the second year of growth after a dip from 2014 to 2015. Overall, federal investment increased by $6.1 billion or 18.4% from 2013 to 2017, but growth has been uneven across federal health agencies. Investment by other sectors, including academic and research institutions, foundations, state and local governments, and voluntary health associations and professional societies, also increased from 2013 to 2017. Looking ahead, medical and health R&D spending is expected to move in an upward trajectory in 2018 but will continue to fall short relative to the health and economic impact of major health threats. -

Clinical Research Services
Clinical Research Services Conducting efficient, innovative clinical research from initial planning to closeout WHY LEIDOS LIFE SCIENCES? In addition to developing large-scale technology programs for U.S. federal f Leidos has expertise agencies with a focus on health, Leidos provides a broad range of clinical supporting all product services and solutions to researchers, product sponsors, U.S. government development phases agencies, and other biomedical enterprises, hospitals, and health systems. for vaccines, drugs, and Our broad research experience enables us to support programs throughout biotherapeutics the development life cycle: from concept through the exploratory, f Leidos understands the development, pre-clinical, and clinical phases, as well as with product need to control clinical manufacturing and launching. Leidos designs and develops customized project scope, timelines, solutions that support groundbreaking medical research, optimize business and cost while remaining operations, and expedite the discovery of safe and effective medical flexible amidst shifting products. We apply our technical knowledge and experience in selecting research requirements institutions, clinical research organizations, and independent research f Leidos remains vendor- facilities to meet the specific needs of each clinical study. We also manage neutral while fostering and team integration, communication, and contracts throughout the project. managing collaborations Finally, Leidos has a proven track record of ensuring that research involving with