Interrogating ARDS Risk and Severity Utilizing Genomic and Systems Biology Approaches
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Nutrigenomics in Human Peripheral Blood Mononuclear Cells
Nutrigenomics in human peripheral blood mononuclear cells The effects of fatty acids on gene expression profiles of human circulating cells as assessed in human intervention studies Mark Bouwens Promotor Prof. dr. Michael R. Müller Hoogleraar Voeding, Metabolisme en Genomics Afdeling Humane Voeding, Wageningen Universiteit Co-promotor Dr. ir. Lydia A. Afman Universitair docent Afdeling Humane Voeding, Wageningen Universiteit Promotiecommissie Prof. dr. Hannelore Daniel Technical University of Munich, Germany Dr. ir. Baukje de Roos Rowett Research Institute, Scotland Prof. dr. ir. Wim H.M. Saris Universiteit Maastricht Prof. dr. Huub F.J. Savelkoul Wageningen Universiteit Dit onderzoek is uitgevoerd binnen de onderzoeksschool VLAG (Voeding, Levensmiddelentechnologie, Agrobiotechnologie en Gezondheid) Nutrigenomics in human peripheral blood mononuclear cells The effects of fatty acids on gene expression profiles of human circulating cells as assessed in human intervention studies Mark Bouwens Proefschrift ter verkrijging van de graad van doctor op gezag van de rector magnificus van Wageningen Universiteit, Prof. dr. M.J. Kropff, in het openbaar te verdedigen op vrijdag 20 februari 2009 des namiddags te half twee in de Aula. Mark Bouwens Nutrigenomics in human peripheral blood mononuclear cells. The effects of fatty acids on gene expression profiles of human circulating cells as assessed in human intervention studies Thesis Wageningen University, Wageningen, the Netherlands, 2009. With abstract – with references – with summary in Dutch. ISBN: 978-90-8585-327-5 Abstract Nutrigenomics in human peripheral blood mononuclear cells. The effects of fatty acids on gene expression profiles of human circulating cells as assessed in human intervention studies PhD thesis by Mark Bouwens, Division of Human Nutrition, Wageningen University, Wageningen, the Netherlands, February 20, 2009 Peripheral blood mononuclear cells (PBMCs) are circulating immune cells. -

WO 2010/129023 A9 11 November 2010 (11.11.2010) PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) CORRECTED VERSION (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2010/129023 A9 11 November 2010 (11.11.2010) PCT (51) International Patent Classification: (74) Agent: BAKER, C , Hunter; Wolf, Greenfield & Sacks, C07K 14/00 (2006.01) P.C., 600 Atlantic Avenue, Boston, MA 02210-2206 (US). (21) International Application Number: PCT/US20 10/00 1250 (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, (22) International Filing Date: AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, 28 April 2010 (28.04.2010) CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, (25) Filing Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, (26) Publication Langi English KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, (30) Priority Data: ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, 61/173,430 28 April 2009 (28.04.2009) US NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, 61/321,428 6 April 2010 (06.04.2010) US SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (71) Applicant (for all designated States except US): PRESI¬ DENT AND FELLOWS OF HARVARD COLLEGE (84) Designated States (unless otherwise indicated, for every [US/US]; 17 Quincy Street, Cambridge, MA 02138 (US). -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

(12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown Et Al
US 20030082511A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown et al. (43) Pub. Date: May 1, 2003 (54) IDENTIFICATION OF MODULATORY Publication Classification MOLECULES USING INDUCIBLE PROMOTERS (51) Int. Cl." ............................... C12O 1/00; C12O 1/68 (52) U.S. Cl. ..................................................... 435/4; 435/6 (76) Inventors: Steven J. Brown, San Diego, CA (US); Damien J. Dunnington, San Diego, CA (US); Imran Clark, San Diego, CA (57) ABSTRACT (US) Correspondence Address: Methods for identifying an ion channel modulator, a target David B. Waller & Associates membrane receptor modulator molecule, and other modula 5677 Oberlin Drive tory molecules are disclosed, as well as cells and vectors for Suit 214 use in those methods. A polynucleotide encoding target is San Diego, CA 92121 (US) provided in a cell under control of an inducible promoter, and candidate modulatory molecules are contacted with the (21) Appl. No.: 09/965,201 cell after induction of the promoter to ascertain whether a change in a measurable physiological parameter occurs as a (22) Filed: Sep. 25, 2001 result of the candidate modulatory molecule. Patent Application Publication May 1, 2003 Sheet 1 of 8 US 2003/0082511 A1 KCNC1 cDNA F.G. 1 Patent Application Publication May 1, 2003 Sheet 2 of 8 US 2003/0082511 A1 49 - -9 G C EH H EH N t R M h so as se W M M MP N FIG.2 Patent Application Publication May 1, 2003 Sheet 3 of 8 US 2003/0082511 A1 FG. 3 Patent Application Publication May 1, 2003 Sheet 4 of 8 US 2003/0082511 A1 KCNC1 ITREXCHO KC 150 mM KC 2000000 so 100 mM induced Uninduced Steady state O 100 200 300 400 500 600 700 Time (seconds) FIG. -

Lipoprotein Lipase: a General Review Moacir Couto De Andrade Júnior1,2*
Review Article iMedPub Journals Insights in Enzyme Research 2018 www.imedpub.com Vol.2 No.1:3 ISSN 2573-4466 DOI: 10.21767/2573-4466.100013 Lipoprotein Lipase: A General Review Moacir Couto de Andrade Júnior1,2* 1Post-Graduation Department, Nilton Lins University, Manaus, Amazonas, Brazil 2Department of Food Technology, Instituto Nacional de Pesquisas da Amazônia (INPA), Manaus, Amazonas, Brazil *Corresponding author: MC Andrade Jr, Post-Graduation Department, Nilton Lins University, Manaus, Amazonas, Brazil, Tel: +55 (92) 3633-8028; E-mail: [email protected] Rec date: March 07, 2018; Acc date: April 10, 2018; Pub date: April 17, 2018 Copyright: © 2018 Andrade Jr MC. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Citation: Andrade Jr MC (2018) Lipoprotein Lipase: A General Review. Insights Enzyme Res Vol.2 No.1:3 Abstract Lipoprotein Lipase: Historical Hallmarks, Enzymatic Activity, Characterization, and Carbohydrates (e.g., glucose) and lipids (e.g., free fatty acids or FFAs) are the most important sources of energy Present Relevance in Human for most organisms, including humans. Lipoprotein lipase (LPL) is an extracellular enzyme (EC 3.1.1.34) that is Pathophysiology and Therapeutics essential in lipoprotein metabolism. LPL is a glycoprotein that is synthesized and secreted in several tissues (e.g., Macheboeuf, in 1929, first described chemical procedures adipose tissue, skeletal muscle, cardiac muscle, and for the isolation of a plasma protein fraction that was very rich macrophages). At the luminal surface of the vascular in lipids but readily soluble in water, such as a lipoprotein [1]. -

Supp Table 6.Pdf
Supplementary Table 6. Processes associated to the 2037 SCL candidate target genes ID Symbol Entrez Gene Name Process NM_178114 AMIGO2 adhesion molecule with Ig-like domain 2 adhesion NM_033474 ARVCF armadillo repeat gene deletes in velocardiofacial syndrome adhesion NM_027060 BTBD9 BTB (POZ) domain containing 9 adhesion NM_001039149 CD226 CD226 molecule adhesion NM_010581 CD47 CD47 molecule adhesion NM_023370 CDH23 cadherin-like 23 adhesion NM_207298 CERCAM cerebral endothelial cell adhesion molecule adhesion NM_021719 CLDN15 claudin 15 adhesion NM_009902 CLDN3 claudin 3 adhesion NM_008779 CNTN3 contactin 3 (plasmacytoma associated) adhesion NM_015734 COL5A1 collagen, type V, alpha 1 adhesion NM_007803 CTTN cortactin adhesion NM_009142 CX3CL1 chemokine (C-X3-C motif) ligand 1 adhesion NM_031174 DSCAM Down syndrome cell adhesion molecule adhesion NM_145158 EMILIN2 elastin microfibril interfacer 2 adhesion NM_001081286 FAT1 FAT tumor suppressor homolog 1 (Drosophila) adhesion NM_001080814 FAT3 FAT tumor suppressor homolog 3 (Drosophila) adhesion NM_153795 FERMT3 fermitin family homolog 3 (Drosophila) adhesion NM_010494 ICAM2 intercellular adhesion molecule 2 adhesion NM_023892 ICAM4 (includes EG:3386) intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)adhesion NM_001001979 MEGF10 multiple EGF-like-domains 10 adhesion NM_172522 MEGF11 multiple EGF-like-domains 11 adhesion NM_010739 MUC13 mucin 13, cell surface associated adhesion NM_013610 NINJ1 ninjurin 1 adhesion NM_016718 NINJ2 ninjurin 2 adhesion NM_172932 NLGN3 neuroligin -

WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T (51) International Patent Classification: David [US/US]; 13539 N . 95th Way, Scottsdale, AZ C12Q 1/68 (2006.01) 85260 (US). (21) International Application Number: (74) Agent: AKHAVAN, Ramin; Caris Science, Inc., 6655 N . PCT/US20 12/0425 19 Macarthur Blvd., Irving, TX 75039 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 14 June 2012 (14.06.2012) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, English (25) Filing Language: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, Publication Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 61/497,895 16 June 201 1 (16.06.201 1) US MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 61/499,138 20 June 201 1 (20.06.201 1) US OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, SD, 61/501,680 27 June 201 1 (27.06.201 1) u s SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, 61/506,019 8 July 201 1(08.07.201 1) u s TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Endogenous Protein Interactome of Human UDP-Glucuronosyltransferases Exposed by Untargeted Proteomics
ORIGINAL RESEARCH published: 03 February 2017 doi: 10.3389/fphar.2017.00023 Endogenous Protein Interactome of Human UDP-Glucuronosyltransferases Exposed by Untargeted Proteomics Michèle Rouleau, Yannick Audet-Delage, Sylvie Desjardins, Mélanie Rouleau, Camille Girard-Bock and Chantal Guillemette * Pharmacogenomics Laboratory, Canada Research Chair in Pharmacogenomics, Faculty of Pharmacy, Centre Hospitalier Universitaire de Québec Research Center, Laval University, Québec, QC, Canada The conjugative metabolism mediated by UDP-glucuronosyltransferase enzymes (UGTs) significantly influences the bioavailability and biological responses of endogenous molecule substrates and xenobiotics including drugs. UGTs participate in the regulation of cellular homeostasis by limiting stress induced by toxic molecules, and by Edited by: controlling hormonal signaling networks. Glucuronidation is highly regulated at genomic, Yuji Ishii, transcriptional, post-transcriptional and post-translational levels. However, the UGT Kyushu University, Japan protein interaction network, which is likely to influence glucuronidation, has received Reviewed by: little attention. We investigated the endogenous protein interactome of human UGT1A Ben Lewis, Flinders University, Australia enzymes in main drug metabolizing non-malignant tissues where UGT expression is Shinichi Ikushiro, most prevalent, using an unbiased proteomics approach. Mass spectrometry analysis Toyama Prefectural University, Japan of affinity-purified UGT1A enzymes and associated protein complexes in liver, -
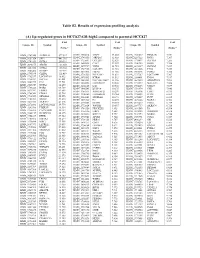
(A) Up-Regulated Genes in HCC827-GR-High2 Compared to Parental HCC827
Table S2. Results of expression profiling analysis (A) Up-regulated genes in HCC827-GR-high2 compared to parental HCC827 Fold Fold Fold Unique ID Symbol Unique ID Symbol Unique ID Symbol change* change* change* ILMN_1709348 ALDH1A1 577.587 ILMN_2310814 MAPT 13.003 ILMN_1741017 PIP4K2B 7.331 ILMN_1651354 SPP1 441.316 ILMN_1748650 MRPL45 12.988 ILMN_3237623 RNY1 7.297 ILMN_1701831 GSTA1 260.591 ILMN_1755897 UGT2B7 12.629 ILMN_1734897 SLC4A4 7.285 ILMN_1658835 CAV2 12.357 ILMN_1746359 RERG 7.280 ILMN_2094875 ABCB1 183.050 ILMN_1678939 VNN2 11.935 ILMN_1671337 SLC2A5 7.257 ILMN_3251540 GSTA2 145.982 ILMN_1729905 GAL3ST1 11.910 ILMN_1691606 LYG2 7.254 ILMN_2062468 IGFBP7 127.721 ILMN_1672536 FBLN1 11.716 ILMN_1785646 PMP22 7.246 ILMN_1795190 CLDN2 111.439 ILMN_1796339 PLEKHA2 11.631 ILMN_1737387 LOC728441 7.207 ILMN_1782937 LOC647169 98.612 ILMN_1676563 HTRA1 11.592 ILMN_1684401 FMO1 7.117 ILMN_1754247 SLC3A1 81.001 ILMN_3263423 LOC100129027 11.346 ILMN_1687035 ADAMTSL4 7.098 ILMN_1662795 CA2 79.581 ILMN_1694898 LOC653857 10.906 ILMN_2153572 MAGEA3 7.086 ILMN_2168747 GSTA2 66.250 ILMN_2404625 LAT 10.560 ILMN_1784283 USH1C 7.079 ILMN_1764228 DAB2 64.709 ILMN_1666546 DUSP14 10.375 ILMN_1731374 CPE 7.046 ILMN_1675797 EPDR1 63.605 ILMN_1764571 ARHGAP23 10.299 ILMN_1765446 EMP3 6.933 ILMN_1708341 PDZK1 59.714 ILMN_3200140 LOC645638 10.284 ILMN_1754002 IL1F8 6.863 ILMN_1713529 SEMA6A 52.575 ILMN_3244343 SNORA21 10.171 ILMN_1878007 FUT9 6.835 ILMN_1708391 NR1H4 43.218 ILMN_1671489 PC 10.075 ILMN_1699208 NAP1L1 6.763 ILMN_2412336 AKR1C2 42.826 ILMN_2404688 -
Figure S1. Reverse Transcription‑Quantitative PCR Analysis of ETV5 Mrna Expression Levels in Parental and ETV5 Stable Transfectants
Figure S1. Reverse transcription‑quantitative PCR analysis of ETV5 mRNA expression levels in parental and ETV5 stable transfectants. (A) Hec1a and Hec1a‑ETV5 EC cell lines; (B) Ishikawa and Ishikawa‑ETV5 EC cell lines. **P<0.005, unpaired Student's t‑test. EC, endometrial cancer; ETV5, ETS variant transcription factor 5. Figure S2. Survival analysis of sample clusters 1‑4. Kaplan Meier graphs for (A) recurrence‑free and (B) overall survival. Survival curves were constructed using the Kaplan‑Meier method, and differences between sample cluster curves were analyzed by log‑rank test. Figure S3. ROC analysis of hub genes. For each gene, ROC curve (left) and mRNA expression levels (right) in control (n=35) and tumor (n=545) samples from The Cancer Genome Atlas Uterine Corpus Endometrioid Cancer cohort are shown. mRNA levels are expressed as Log2(x+1), where ‘x’ is the RSEM normalized expression value. ROC, receiver operating characteristic. Table SI. Clinicopathological characteristics of the GSE17025 dataset. Characteristic n % Atrophic endometrium 12 (postmenopausal) (Control group) Tumor stage I 91 100 Histology Endometrioid adenocarcinoma 79 86.81 Papillary serous 12 13.19 Histological grade Grade 1 30 32.97 Grade 2 36 39.56 Grade 3 25 27.47 Myometrial invasiona Superficial (<50%) 67 74.44 Deep (>50%) 23 25.56 aMyometrial invasion information was available for 90 of 91 tumor samples. Table SII. Clinicopathological characteristics of The Cancer Genome Atlas Uterine Corpus Endometrioid Cancer dataset. Characteristic n % Solid tissue normal 16 Tumor samples Stagea I 226 68.278 II 19 5.740 III 70 21.148 IV 16 4.834 Histology Endometrioid 271 81.381 Mixed 10 3.003 Serous 52 15.616 Histological grade Grade 1 78 23.423 Grade 2 91 27.327 Grade 3 164 49.249 Molecular subtypeb POLE 17 7.328 MSI 65 28.017 CN Low 90 38.793 CN High 60 25.862 CN, copy number; MSI, microsatellite instability; POLE, DNA polymerase ε. -

Resveratrol Inhibits Cell Cycle Progression by Targeting Aurora Kinase a and Polo-Like Kinase 1 in Breast Cancer Cells
3696 ONCOLOGY REPORTS 35: 3696-3704, 2016 Resveratrol inhibits cell cycle progression by targeting Aurora kinase A and Polo-like kinase 1 in breast cancer cells RUBICELI MEDINA-AGUILAR1, LAURENCE A. Marchat2, ELENA ARECHAGA OCAMPO3, Patricio GARIGLIO1, JAIME GARCÍA MENA1, NICOLÁS VILLEGAS SEPÚlveda4, MACARIO MartÍNEZ CASTILLO4 and CÉSAR LÓPEZ-CAMARILLO5 1Department of Genetics and Molecular Biology, CINVESTAV-IPN, Mexico D.F.; 2Molecular Biomedicine Program and Biotechnology Network, National School of Medicine and Homeopathy, National Polytechnic Institute, Mexico D.F.; 3Natural Sciences Department, Metropolitan Autonomous University, Mexico D.F.; 4Department of Molecular Biomedicine, CINVESTAV-IPN, Mexico D.F.; 5Oncogenomics and Cancer Proteomics Laboratory, Universidad Autónoma de la Ciudad de México, Mexico D.F., Mexico Received December 4, 2015; Accepted January 8, 2016 DOI: 10.3892/or.2016.4728 Abstract. The Aurora protein kinase (AURKA) and the MDA-MB-231 and MCF-7 cells. By western blot assays, we Polo-like kinase-1 (PLK1) activate the cell cycle, and they confirmed that resveratrol suppressed AURKA, CCND1 and are considered promising druggable targets in cancer CCNB1 at 24 and 48 h. In summary, we showed for the first time therapy. However, resistance to chemotherapy and to specific that resveratrol regulates cell cycle progression by targeting small-molecule inhibitors is common in cancer patients; thus AURKA and PLK1. Our findings highlight the potential use of alternative therapeutic approaches are needed to overcome resveratrol as an adjuvant therapy for breast cancer. clinical resistance. Here, we showed that the dietary compound resveratrol suppressed the cell cycle by targeting AURKA Introduction and PLK1 kinases. First, we identified genes modulated by resveratrol using a genome-wide analysis of gene expression Cancer development results from the interaction between in MDA-MB-231 breast cancer cells.