Recognition of Tumors by the Innate Immune System and Natural Killer Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Downloaded in Aug 2019
ARTICLE https://doi.org/10.1038/s41467-021-24473-2 OPEN IL-15 and PIM kinases direct the metabolic programming of intestinal intraepithelial lymphocytes Olivia J. James1, Maud Vandereyken1, Julia M. Marchingo 2, Francois Singh1, Susan E. Bray3, Jamie Wilson4, ✉ Andrew G. Love1 & Mahima Swamy 1 1234567890():,; Intestinal intraepithelial lymphocytes (IEL) are an abundant population of tissue-resident T cells that protect and maintain the intestinal barrier. IEL respond to epithelial cell-derived IL-15, which is complexed to the IL-15 receptor α chain (IL-15/Rα). IL-15 is essential both for maintaining IEL homeostasis and inducing IEL responses to epithelial stress, which has been associated with Coeliac disease. Here, we apply quantitative mass spectrometry to IL-15/Rα- stimulated IEL to investigate how IL-15 directly regulates inflammatory functions of IEL. IL-15/ Rα drives IEL activation through cell cycle regulation, upregulation of metabolic machinery and expression of a select repertoire of cell surface receptors. IL-15/Rα selectively upregu- lates the Ser/Thr kinases PIM1 and PIM2, which are essential for IEL to proliferate, grow and upregulate granzyme B in response to inflammatory IL-15. Notably, IEL from patients with Coeliac disease have high PIM expression. Together, these data indicate PIM kinases as important effectors of IEL responses to inflammatory IL-15. 1 MRC Protein Phosphorylation and Ubiquitylation Unit, University of Dundee, Dundee, UK. 2 Division of Cell Signalling and Immunology, School of Life Sciences, University of Dundee, Dundee, UK. 3 NHS Research Scotland, Tayside Tissue Biorepository, University of Dundee, Dundee, UK. 4 Department of ✉ Pathology, NHS Tayside, Ninewells Hospital, Dundee, UK. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Supplementary Table S5. Differentially Expressed Gene Lists of PD-1High CD39+ CD8 Tils According to 4-1BB Expression Compared to PD-1+ CD39- CD8 Tils
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Immunother Cancer Supplementary Table S5. Differentially expressed gene lists of PD-1high CD39+ CD8 TILs according to 4-1BB expression compared to PD-1+ CD39- CD8 TILs Up- or down- regulated genes in Up- or down- regulated genes Up- or down- regulated genes only PD-1high CD39+ CD8 TILs only in 4-1BBneg PD-1high CD39+ in 4-1BBpos PD-1high CD39+ CD8 compared to PD-1+ CD39- CD8 CD8 TILs compared to PD-1+ TILs compared to PD-1+ CD39- TILs CD39- CD8 TILs CD8 TILs IL7R KLRG1 TNFSF4 ENTPD1 DHRS3 LEF1 ITGA5 MKI67 PZP KLF3 RYR2 SIK1B ANK3 LYST PPP1R3B ETV1 ADAM28 H2AC13 CCR7 GFOD1 RASGRP2 ITGAX MAST4 RAD51AP1 MYO1E CLCF1 NEBL S1PR5 VCL MPP7 MS4A6A PHLDB1 GFPT2 TNF RPL3 SPRY4 VCAM1 B4GALT5 TIPARP TNS3 PDCD1 POLQ AKAP5 IL6ST LY9 PLXND1 PLEKHA1 NEU1 DGKH SPRY2 PLEKHG3 IKZF4 MTX3 PARK7 ATP8B4 SYT11 PTGER4 SORL1 RAB11FIP5 BRCA1 MAP4K3 NCR1 CCR4 S1PR1 PDE8A IFIT2 EPHA4 ARHGEF12 PAICS PELI2 LAT2 GPRASP1 TTN RPLP0 IL4I1 AUTS2 RPS3 CDCA3 NHS LONRF2 CDC42EP3 SLCO3A1 RRM2 ADAMTSL4 INPP5F ARHGAP31 ESCO2 ADRB2 CSF1 WDHD1 GOLIM4 CDK5RAP1 CD69 GLUL HJURP SHC4 GNLY TTC9 HELLS DPP4 IL23A PITPNC1 TOX ARHGEF9 EXO1 SLC4A4 CKAP4 CARMIL3 NHSL2 DZIP3 GINS1 FUT8 UBASH3B CDCA5 PDE7B SOGA1 CDC45 NR3C2 TRIB1 KIF14 TRAF5 LIMS1 PPP1R2C TNFRSF9 KLRC2 POLA1 CD80 ATP10D CDCA8 SETD7 IER2 PATL2 CCDC141 CD84 HSPA6 CYB561 MPHOSPH9 CLSPN KLRC1 PTMS SCML4 ZBTB10 CCL3 CA5B PIP5K1B WNT9A CCNH GEM IL18RAP GGH SARDH B3GNT7 C13orf46 SBF2 IKZF3 ZMAT1 TCF7 NECTIN1 H3C7 FOS PAG1 HECA SLC4A10 SLC35G2 PER1 P2RY1 NFKBIA WDR76 PLAUR KDM1A H1-5 TSHZ2 FAM102B HMMR GPR132 CCRL2 PARP8 A2M ST8SIA1 NUF2 IL5RA RBPMS UBE2T USP53 EEF1A1 PLAC8 LGR6 TMEM123 NEK2 SNAP47 PTGIS SH2B3 P2RY8 S100PBP PLEKHA7 CLNK CRIM1 MGAT5 YBX3 TP53INP1 DTL CFH FEZ1 MYB FRMD4B TSPAN5 STIL ITGA2 GOLGA6L10 MYBL2 AHI1 CAND2 GZMB RBPJ PELI1 HSPA1B KCNK5 GOLGA6L9 TICRR TPRG1 UBE2C AURKA Leem G, et al. -

32-13307: LY9 Human Description Product Info
9853 Pacific Heights Blvd. Suite D. San Diego, CA 92121, USA Tel: 858-263-4982 Email: [email protected] 32-13307: LY9 Human Lymphocyte Antigen 9, Signaling Lymphocytic Activation Molecule 3, Cell Surface Molecule Ly-9, SLAM Alternative Name : Family Member 3, SLAMF3, CD229 Antigen, CD229, Hly9, MLY9, LY9. Description Source: Sf9, Baculovirus cells. Sterile filtered colorless solution. Lymphocyte Antigen 9, also known as LY9 is a member of the SLAM family of immunomodulatory receptors, LY9 interacts with the adaptor molecule SAP . LY9 plays a part in adhesion reactions between T lymphocytes and accessory cells via homophilic interaction. LY9 promotes T-cell differentiation into a helper T-cell Th17 phenotype which leads to increased IL-17 secretion; the costimulatory activity requires SH2D1A. Furthermore it also promotes recruitment of RORC to the IL-17 promoter. LY9 is implicated in the maintenance of peripheral cell tolerance through serving as a negative regulator of the immune response. LY9 disables autoantibody responses as well as inhibits IFN-gamma secretion by CD4(+) T-cells. Moreover, LY9 negatively regulate the size of thymic innate CD8(+) T-cells and the development of invariant natural killer T (iNKT) cells. Systemic Lupus Erythematosus is one of the diseases which are associated with LY9. LY9 Human Recombinant produced in Sf9 Baculovirus cells is a single, glycosylated polypeptide chain containing 416 amino acids (48-454 aa) and having a molecular mass of 45.9kDa (Migrates at 40-57kDa on SDS-PAGE under reducing conditions).LY9 is expressed with a 6 amino acid His tag at C-Terminus and purified by proprietary chromatographic techniques. -

Characteristics of B Cell-Associated Gene Expression in Patients With
MOLECULAR MEDICINE REPORTS 13: 4113-4121, 2016 Characteristics of B cell-associated gene expression in patients with coronary artery disease WENWEN YAN*, HAOMING SONG*, JINFA JIANG, WENJUN XU, ZHU GONG, QIANGLIN DUAN, CHUANGRONG LI, YUAN XIE and LEMIN WANG Department of Internal Medicine, Division of Cardiology, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, P.R. China Received May 19, 2015; Accepted February 12, 2016 DOI: 10.3892/mmr.2016.5029 Abstract. The current study aimed to identify differentially with the two other groups. Additionally the gene expression expressed B cell-associated genes in peripheral blood mono- levels of B cell regulatory genes were measured. In patients nuclear cells and observe the changes in B cell activation at with AMI, CR1, LILRB2, LILRB3 and VAV1 mRNA expres- different stages of coronary artery disease. Groups of patients sion levels were statistically increased, whereas, CS1 and IL4I1 with acute myocardial infarction (AMI) and stable angina (SA), mRNAs were significantly reduced compared with the SA and as well as healthy volunteers, were recruited into the study control groups. There was no statistically significant difference (n=20 per group). Whole human genome microarray analysis in B cell-associated gene expression levels between patients was performed to examine the expression of B cell-associated with SA and the control group. The present study identified the genes among these three groups. The mRNA expression levels downregulation of genes associated with BCRs, B2 cells and of 60 genes associated with B cell activity and regulation were B cell regulators in patients with AMI, indicating a weakened measured using reverse transcription-quantitative polymerase T cell-B cell interaction and reduced B2 cell activation during chain reaction. -
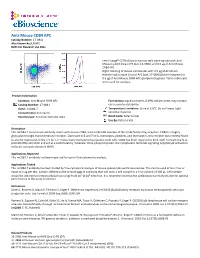
Anti-Mouse CD84 APC Catalog Number: 17‐0841 Also Known As:SLAMF5 RUO: for Research Use Only
Anti-Mouse CD84 APC Catalog Number: 17‐0841 Also Known As:SLAMF5 RUO: For Research Use Only Left: Lineagelo C57BL/6 bone marrow cells were stained with Anti‐ Mouse Ly‐6A/E (Sca‐1) PE (cat. 12‐5981) and 0.5 µg of Anti‐Mouse CD84 APC. Right: Staining of mouse splenocytes with 0.5 µg of Armenian Hamster IgG Isotype Control APC (cat. 17‐4888) (blue histogram) or 0.5 µg of Anti‐Mouse CD84 APC (purple histogram). Total viable cells were used for analysis. Product Information Contents: Anti‐Mouse CD84 APC Formulation: aqueous buffer, 0.09% sodium azide, may contain Catalog Number: 17‐0841 carrier protein/stabilizer Clone: mCD84.7 Temperature Limitation: Store at 2‐8°C. Do not freeze. Light Concentration: 0.2 mg/mL sensitive material. Host/Isotype: Armenian Hamster IgG1 Batch Code: Refer to Vial Use By: Refer to Vial Description This mCD84.7 monoclonal antibody reacts with mouse CD84, a 64‐ to 82‐kDa member of the SLAM family of Ig receptors. CD84 is a highly glycosylated single transmembrane receptor. Expressed in B and T cells, monocytes, platelets, and thymocytes, this receptor was recently found to also be expressed on Sca‐1+c‐kit+Lin‐ mouse bone marrow hematopoietic stem cells. CD84 has been reported to bind itself homophilically to promote IFNγ secretion and act as a costimulatory molecule. Once phosphorylated, the cytoplasmic tail binds signaling lymphocyte activation molecule‐associated protein (SAP). Applications Reported This mCD84.7 antibody has been reported for use in flow cytometric analysis. Applications Tested This mCD84.7 antibody has been tested by flow cytometric analysis of mouse splenocytes and bone marrow. -

Single Cell RNA-Seq and Mass Cytometry Reveals a Novel and a Targetable Population Of
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.04.425268; this version posted January 5, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Single Cell RNA-seq and Mass Cytometry Reveals a Novel and a Targetable Population of Macrophages in Idiopathic Pulmonary Fibrosis Authors: 5 Ayaub EA4*, Poli S2*, Ng J4, Adams T3, Schupp J3, Quesada-Arias L2, Poli F1, Cosme C3, Robertson M6 , Martinez-Manzano J4, Liang X1,4, Villalba J5, Lederer J4, Chu SG4, Raby BA4, Washko G4, Coarfa C6, Perrella MA4, El-Chemaly S4, Kaminski N3, Rosas IO1 Author Affiliations: 10 1 Pulmonary, Critical Care and Sleep Medicine, Baylor College of Medicine, Houston, TX, USA 2 Mount Sinai Medical Center, Division of Internal Medicine, Miami Beach, FL, USA. 3 Pulmonary, Critical Care and Sleep Medicine, Yale School of Medicine, New Haven, CT, USA 4Division of Pulmonary and Critical Care Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA 15 5 Department of Pathology, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts, USA. 6 Dan L Duncan Comprehensive Cancer Center, Baylor College of Medicine, Houston, Texas, USA 20 *These authors contributed equally to the work 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.01.04.425268; this version posted January 5, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Author Contributions: EA, SP, IOR, and NK conceptualized the study. -

Impaired T Lymphocyte Function Increases Tumorigenicity and Decreases Tumor Latency in a Mouse Model of Head and Neck Cancer
1211-1221.qxd 8/9/2009 09:41 Ì ™ÂÏ›‰·1211 INTERNATIONAL JOURNAL OF ONCOLOGY 35: 1211-1221, 2009 Impaired T lymphocyte function increases tumorigenicity and decreases tumor latency in a mouse model of head and neck cancer TONY K.S. KU and DAVID L. CROWE University of Illinois Cancer Center, Chicago, IL 60612, USA Received June 15, 2009; Accepted August 10, 2009 DOI: 10.3892/ijo_00000438 Abstract. Squamous cell carcinoma of the head and neck nations, comprising up to 50% of all malignant tumors. SCC (HNSCC) is the sixth most frequent cancer worldwide. SCC is the most common malignant tumor of the oral cavity with is the most common malignant tumor of the oral cavity with over 35,000 cases and 8,000 deaths reported in the United over 35,000 cases and 8,000 deaths reported in the United States each year (2). Tobacco carcinogens are the primary States each year. Previous case studies have reported increased etiologic agents of the disease with age and genetic background incidence of HNSCC in patients on immunosuppressive as contributory factors. The overall 5-year survival rate is low therapy for organ transplantation. The results of these studies among the major cancers and has not declined significantly in indicate that effective immune surveillance is important for recent years. preventing emergence of HNSCC. HNSCC may also inhibit Previous case studies have reported increased incidence immune response to tumor cells, which may be responsible for of HNSCC in patients on immunosuppressive therapy for progression. We previously reported induction of metastatic organ transplantation (3-7). -

CD84 Cell Surface Signaling Molecule an Emerging Biomarker
Clinical Immunology 204 (2019) 43–49 Contents lists available at ScienceDirect Clinical Immunology journal homepage: www.elsevier.com/locate/yclim Review Article CD84 cell surface signaling molecule: An emerging biomarker and target for cancer and autoimmune disorders T ⁎ Marta Cuencaa, , Jordi Sintesa, Árpád Lányib, Pablo Engela,c a Immunology Unit, Department of Biomedical Sciences, Faculty of Medicine, University of Barcelona, Barcelona, Spain b Department of Immunology, Faculty of Medicine, University of Debrecen, Debrecen, Hungary c Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain ARTICLE INFO ABSTRACT Keywords: CD84 (SLAMF5) is a member of the SLAM family of cell-surface immunoreceptors. Broadly expressed on most CD84 immune cell subsets, CD84 functions as a homophilic adhesion molecule, whose signaling can activate or inhibit SLAM family receptors leukocyte function depending on the cell type and its stage of activation or differentiation. CD84-mediated Germinal center (GC) signaling regulates diverse immunological processes, including T cell cytokine secretion, natural killer cell cy- Autophagy totoxicity, monocyte activation, autophagy, cognate T:B interactions, and B cell tolerance at the germinal center Disease biomarkers checkpoint. Recently, alterations in CD84 have been related to autoimmune and lymphoproliferative disorders. Autoimmune disorders fi Chronic lymphocytic leukemia (CLL) Speci c allelic variations in CD84 are associated with autoimmune diseases such as systemic lupus er- ythematosus and rheumatoid arthritis. In chronic lymphocytic leukemia, CD84 mediates intrinsic and stroma- induced survival of malignant cells. In this review, we describe our current understanding of the structure and function of CD84 and its potential role as a therapeutic target and biomarker in inflammatory autoimmune disorders and cancer. -

Studies on the Function and Regulation of CD84, GPVI and Orai2 in Genetically Modified Mice
Julius-Maximilians-Universität Würzburg Studies on the function and regulation of CD84, GPVI and Orai2 in genetically modified mice Untersuchungen zur Funktion und Regulation von CD84, GPVI und Orai2 in genetisch veränderten Mäusen Doctoral thesis for a doctoral degree at the Graduate School of Life Sciences Section Biomedicine submitted by Sebastian Hofmann from Coburg Würzburg, 2013 Submitted on: ………………………………………………………………………………… Office stamp Members of the Promotionskomitee: Chairperson: Prof. Dr. Manfred Gessler Primary Supervisor: Prof. Dr. Bernhard Nieswandt Supervisor (Second): PD Dr. Heike Hermanns Supervisor (Third): Prof. Dr. Guido Stoll Date of Public Defence: ……………………………………………………………………… Date of Receipt of Certificates: ……………………………………………………………... Summary Summary Platelet activation and aggregation at sites of vascular injury are essential processes to limit blood loss but they also contribute to arterial thrombosis, which can lead to myocardial infarction and stroke. Stable thrombus formation requires a series of events involving platelet receptors which contribute to adhesion, activation and aggregation of platelets. Regulation of receptor expression by (metallo-)proteinases has been described for several platelet receptors, but the molecular mechanisms are ill-defined. The signaling lymphocyte activation molecule (SLAM) family member CD84 is expressed in immune cells and platelets, however its role in platelet physiology was unclear. In this thesis, CD84 deficient mice were generated and analyzed. In well established in vitro and in vivo assays testing platelet function and thrombus formation, CD84 deficient mice displayed phenotypes indistinguishable from wild-type controls. It was concluded that CD84 in platelets does not function as modulator of thrombus formation, but rather has other functions. In line with this, in the second part of this thesis, a novel regulation mechanism for platelet CD84 was discovered and elucidated.