Polymerization Catalysts with Dn Electrons (N ) 1-4): a Theoretical Study
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

5.111 Principles of Chemical Science, Fall 2005 Transcript – Lecture 28
MIT OpenCourseWare http://ocw.mit.edu 5.111 Principles of Chemical Science, Fall 2005 Please use the following citation format: Sylvia Ceyer and Catherine Drennan, 5.111 Principles of Chemical Science, Fall 2005. (Massachusetts Institute of Technology: MIT OpenCourseWare). http://ocw.mit.edu (accessed MM DD, YYYY). License: Creative Commons Attribution-Noncommercial-Share Alike. Note: Please use the actual date you accessed this material in your citation. For more information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms MIT OpenCourseWare http://ocw.mit.edu 5.111 Principles of Chemical Science, Fall 2005 Transcript – Lecture 28 All right. We have a few more PowerPoint things before I am going to attempt using the board. Again, we are in the transition metal unit. And today we are going to introduce something called crystal field theory. And this is in Chapter 16 in your book. Let me just tell you about two different theories. Again, chemistry is an experimental science. We collect data and then we try to come up with theories that explain the data, so the theories are not sort of 100%. And some are more simple and some are more complicated to try to explain what we observe. And some of these theories, although they are pretty simple approximations of what is really going on, do a pretty good job of explaining the things that we are observing. All right. There are two different kinds of theories that you will hear about with transition metals. You will hear about crystal field theory and ligand field theory. -

Organometrallic Chemistry
CHE 425: ORGANOMETALLIC CHEMISTRY SOURCE: OPEN ACCESS FROM INTERNET; Striver and Atkins Inorganic Chemistry Lecturer: Prof. O. G. Adeyemi ORGANOMETALLIC CHEMISTRY Definitions: Organometallic compounds are compounds that possess one or more metal-carbon bond. The bond must be “ionic or covalent, localized or delocalized between one or more carbon atoms of an organic group or molecule and a transition, lanthanide, actinide, or main group metal atom.” Organometallic chemistry is often described as a bridge between organic and inorganic chemistry. Organometallic compounds are very important in the chemical industry, as a number of them are used as industrial catalysts and as a route to synthesizing drugs that would not have been possible using purely organic synthetic routes. Coordinative unsaturation is a term used to describe a complex that has one or more open coordination sites where another ligand can be accommodated. Coordinative unsaturation is a very important concept in organotrasition metal chemistry. Hapticity of a ligand is the number of atoms that are directly bonded to the metal centre. Hapticity is denoted with a Greek letter η (eta) and the number of bonds a ligand has with a metal centre is indicated as a superscript, thus η1, η2, η3, ηn for hapticity 1, 2, 3, and n respectively. Bridging ligands are normally preceded by μ, with a subscript to indicate the number of metal centres it bridges, e.g. μ2–CO for a CO that bridges two metal centres. Ambidentate ligands are polydentate ligands that can coordinate to the metal centre through one or more atoms. – – – For example CN can coordinate via C or N; SCN via S or N; NO2 via N or N. -

Organometallic and Catalysis
ORGANOMETALLIC AND CATALYSIS Dr. Malay Dolai, Assistant Professor, Department of Chemistry, Prabhat Kumar College, Contai, Purba Medinipur-721404, WB, India. 1.Introduction Organometallic chemistry is the study of organometallic compounds, chemical compounds containing at least one chemical bond between a carbon atom of an organic molecule and a metal, including alkaline, alkaline earth, and transition metals, and sometimes broadened to include metalloids like boron, silicon, and tin, as well. Aside from bonds to organyl fragments or molecules, bonds to 'inorganic' carbon, like carbon monoxide (metal carbonyls), cyanide, or carbide, are generally considered to be organometallic as well. Some related compounds such as transition metal hydrides and metal phosphine complexes are often included in discussions of organometallic compounds, though strictly speaking, they are not necessarily organometallic. The related but distinct term "metalorganic compound" refers to metal-containing compounds lacking direct metal-carbon bonds but which contain organic ligands. In 1827, Zeise's salt is the first platinum- olefin complex: K[PtCl3(C2H4)].H2O, the first invented organometallic compound. Organometallic compounds find wide use in commercial reactions, both as homogeneous catalysis and as stoichiometric reagents For instance, organolithium, organomagnesium, and organoaluminium compounds, examples of which are highly basic and highly reducing, are useful stoichiometrically, but also catalyze many polymerization reactions. Almost all processes involving carbon monoxide rely on catalysts, notable examples being described as carbonylations. The production of acetic acid from methanol and carbon monoxide is catalyzed via metal carbonyl complexes in the Monsanto process and Cativa process. Most synthetic aldehydes are produced via hydroformylation. The bulk of the synthetic alcohols, at least those larger than ethanol, are produced by hydrogenation of hydroformylation- derived aldehydes. -

Metal-Ligand Bonding and Inorganic Reaction Mechanisms Year 2
Metal-Ligand Bonding and Inorganic Reaction Mechanisms Year 2 RED Metal-ligand and metal-metal bonding of the transition metal elements Synopsis Lecture 1: Trends of the transition metal series. Ionic vs Covalent bonding. Nomenclature. Electron counting. Lecture 2: Thermodynamics of complex formation. Why complexes form. Recap of molecular orbital theory. 18-electron rule. Lecture 3: Ligand classes. -donor complexes. Octahedral ML6 molecular orbital energy diagram. Lecture 3: - acceptor ligands and synergic bonding. Binding of CO, CN , N2, O2 and NO. Lecture 4: Alkenes, M(H2) vs M(H)2, Mn(O2) complexes, PR3. Lecture 5: 2- - 2- 3- donor ligands, metal-ligand multiple bonds, O , R2N , RN , N . Lecture 6: ML6 molecular orbital energy diagrams incorporating acceptor and donor ligands. Electron counting revisited and link to spectrochemical series. Lecture 7: Kinetics of complex formation. Substitution mechanisms of inorganic complexes. Isomerisation. Lecture 8: Ligand effects on substitution rates (trans-effect, trans-influence). Metal and geometry effects on substitution rates. Lecture 9: Outer sphere electron transfer. Lecture 10: Inner Sphere electron transfer. Bridging ligands. 2 Learning Objectives: by the end of the course you should be able to i) Use common nomenclature in transition metal chemistry. ii) Count valence electrons and determine metal oxidation state in transition metal complexes. iii) Understand the physical basis of the 18-electron rule. iv) Appreciate the synergic nature of bonding in metal carbonyl complexes. v) Understand the relationship between CO, the 'classic' -acceptor and related ligands such as NO, CN, N2, and alkenes. 2 vi) Describe the nature of the interaction between -bound diatomic molecules (H2, O2) and their relationship to -acceptor ligands. -

Materials Sciences Programs Fiscal Year 1994
DOE/ER-0648 Dist. Category UC-404 April 1995 Materials Sciences Programs Fiscal Year 1994 U.S. Department of Energy Office ofEnergy Research Office of Basic Energy Sciences Division of Materials Sciences Germantown Building 19901 Germantown Road Germantown, MD 20874-1290 FOREWORD The Division of Materials Sciences is located within the Department of Energy (DOE) in the Office of Basic Energy Sciences which is under the Office of Energy Research. The Director of the Office of Energy Research is appointed by the President and confirmed by the Senate. The Director of the Office of Energy Research is responsible for oversight of, and providing advice to, the Secretary of Energy on the Department's research portfolio and on the management of all of the Laboratories that It owns, except for those that are designated as having a primary role in nuclear weaponry. The Division of Materials Sciences Is responsible for basic research and research facilities in strategic materials science topics of critical importance to the mission of the Department and its Strategic Plan. Other programmatic divisions under the Office of Basic Energy Sciences are Chemical Sciences, Engineering and Geosciences, and Energy Biosciences; information for them is contained on page 165. Materials Science is an enabling technology. The performance parameters, economics, environmental acceptability and safety of all energy generation, conversion, transmission and conservation technologies are limited by the properties and behavior of materials. The Materials Sciences programs develop scientific understanding of the synergistic relationship amongst the synthesis, processing, structure, properties, behavior, performance and other characteristics of materials. Emphasis is placed on the development of the capability to discover technologically, economically, and environmentally desirable new materials and processes, and the instruments and national user facilities necessary for achieving such progress. -

Metal–Dithiolene Bonding Contributions to Pyranopterin Molybdenum Enzyme Reactivity
inorganics Review Metal–Dithiolene Bonding Contributions to Pyranopterin Molybdenum Enzyme Reactivity Jing Yang 1 , John H. Enemark 2 and Martin L. Kirk 1,* 1 Department of Chemistry and Chemical Biology, The University of New Mexico, MSC03 2060, Albuquerque, NM 87131-0001, USA; [email protected] 2 Department of Chemistry Biochemistry, University of Arizona, Tucson, AZ 85721, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-505-277-5992 Received: 2 February 2020; Accepted: 2 March 2020; Published: 5 March 2020 Abstract: Here we highlight past work on metal–dithiolene interactions and how the unique electronic structure of the metal–dithiolene unit contributes to both the oxidative and reductive half reactions in pyranopterin molybdenum and tungsten enzymes. The metallodithiolene electronic structures detailed here were interrogated using multiple ground and excited state spectroscopic probes on the enzymes and their small molecule analogs. The spectroscopic results have been interpreted in the context of bonding and spectroscopic calculations, and the pseudo-Jahn–Teller effect. The dithiolene is a unique ligand with respect to its redox active nature, electronic synergy with the pyranopterin component of the molybdenum cofactor, and the ability to undergo chelate ring distortions that control covalency, reduction potential, and reactivity in pyranopterin molybdenum and tungsten enzymes. Keywords: metal–dithiolene; pyranopterin molybdenum enzymes; fold-angle; tungsten enzymes; electronic structure; pseudo-Jahn–Teller effect; thione; molybdenum cofactor; Moco 1. Introduction It is now well-established that all known molybdenum-containing enzymes [1–3], with the sole exception of nitrogenase, contain a common pyranopterin dithiolene (PDT) (Figure1) organic cofactor (originally called molybdopterin (MPT)), which coordinates to the Mo center of the enzymes through the sulfur atoms of the dithiolene fragment. -

21.1-21.4, 22 Ch
Assigned Reading: 21.1-21.4, 22 Ch 102 – Problem Set 6 Due : Thursday, May 20 – Before Class Problem 1 (20 pts) For the following molecules, (i) Determine the oxidation state and d electron count of the metal (ii) Identify the compound as high spin or low spin (iii) Calculate the spin-only magnetic moment based on the number of unpaired electrons 3- a) [FeBr6] 2+ b) [Co(NH3)6] 3+ c) [Co(NH3)6] 3+ d) [Mn(CO)6] 3- e) [OsBr6] 2- f) [RhF6] g) TiCl4 - h) [TiCl4] Problem 2 (20 pts) For the following transition metal complexes, propose the most reasonable structure. Keep in mind the 18-electron rule. Give the valence shell electron count, the formal oxidation state and the d-electron count. Make sure that the ligand hapticity is clear in your drawings. Indicate clearly whether the NO ligand is bent or linear, if applicable. a. (C6H6)Fe(CO)2PPh3 2+ b. [(C6H6)Fe(CO)2PPh3] c. Fe(NO)2(CO)2 d. (C6Me6)2Ru (C6Me6 = hexamethylbenzene) e. Mn2(CO)10 f. [(C6H6)W(C3H5)]2(µ-Cl)2 + g. [CoCl(en)2(NO)] (en = ethane-1,2-diamine, a.k.a. ethylenediamine) Problem 3 (15 Points) For the following transition metal complexes, give the valence shell electron count, the formal oxidation state, and the d-electron count. a. PR2 Ph B Fe N R = H2C P R 2PR2 b. tBuO OtBu OtBu W W tBuO tBuO OtBu c. (H3C)2HC Cl Cl CH(CH3)2 (H C) HC CH(CH ) 3 2 P Ni Ni P 3 2 d. -
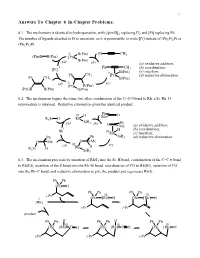
Answers to Chapter 6 In-Chapter Problems
1 Answers To Chapter 6 In-Chapter Problems. 6.1. The mechanism is identical to hydrogenation, with [(pin)B]2 replacing H2 and [Pt] replacing Pd. The number of ligands attached to Pt is uncertain, so it is permissible to write [Pt] instead of (Ph3P)2Pt or (Ph3P)3Pt. II B(Pin) Ph CH3 (Pin)B B(Pin) [Pt] B(Pin) (a) (b) (a) oxidative addition; 0 Ph CH (b) coordination; [Pt] 3 II B(Pin) (c) insertion; Ph CH3 [Pt] (d) reductive elimination. Ph CH3 B(Pin) (d) II (c) [Pt] B(Pin) (Pin)B B(Pin) B(Pin) 6.2. The mechanism begins the same, but after coordination of the C=O π bond to Rh, a Si–Rh–O intermediate is obtained. Reductive elimination gives the identical product. Ph III H Me O R3Si H Rh (a) SiR3 (b) Ph O Me (a) oxidative addition; I H (b) coordination; Rh IIIRh (c) insertion; Ph Ph SiR3 (d) reductive elimination. O Me O Me III (c) (d) Rh H R3Si H SiR3 6.3. The mechanism proceeds by insertion of Rh(I) into the Si–H bond, coordination of the C=C π bond to Rh(III), insertion of the π bond into the Rh–Si bond, coordination of CO to Rh(III), insertion of CO into the Rh–C bond, and reductive elimination to give the product and regenerate Rh(I). Ph Ph OSi H Ph Ph Ph Ph i-Pr III III I OSi [Rh] H OSi [Rh] H [Rh] i-Pr i-Pr product Ph Ph H Ph Ph H Ph Ph III III III OSi [Rh] C O OSi [Rh] C O OSi [Rh] H i-Pr i-Pr i-Pr Chapter 6 2 6.4. -

The 18 Electron Guideline: a Primmer This Can Be Given to Students to Read
Created by Burke Scott Williams ([email protected]) and posted on VIPEr on 01/10/2009. This work is licensed under the Creative Commons Attribution Non-commercial Share Alike License in 2009. To view a copy of this license visit http://creativecommons.org/about/license/ The 18 Electron Guideline: A Primmer This can be given to students to read The 18 electron “rule” is a very useful tool for predicting which complexes will behave as “closed shell” or “saturated” species. Generally speaking, transition metal compounds with more or fewer than 18 valence electrons will be less stable than close analogs that do have 18 electrons. The 18 electron “rule” (s orbital, 3 p-orbitals, 5 d-orbitals, 2 e- in each) ( is a close analog to the “octet rule” (1 s orbital, 3 p-orbitals, 2 e- in each) in general and organic chemistry, but the much larger number of exceptions in transition metal chemistry is such that it’s really more of a “guideline” than a “rule”. But how do we learn to count to 18? There are two primary methods (somewhat analogous to the “oxidation state/ionic model” and the “formal charge/covalent model” in general chemistry. In these more traditional models, we can count the electrons around carbon in CO2 in one of two ways: Oxidation State method: 1. Remove “ligands” (oxygen) as closed shell (full octet) species, and assign oxidation states to the resultant cations and anions: C4+ + 2 O2- 2. Now bring the “ligands” back in. Each bond is worth two electrons (4 for double bonds, 6 for triple) 2 double bonds = 8 electrons, carbon is an “8 electron species” Formal Charge method: 1. -

Electron Counting, Formal Oxidation States and the 18 Electron Rule in Transition Metal Chemistry
Electron Counting, Formal Oxidation States and the 18 Electron Rule in Transition Metal Chemistry The 18 Electron Rule In 1927 Sidgwick proposed that the coordinate bond arises from donation of an electron pair from the ligand to the metal ion. Since noble gases are inert (relatively !!) and have filled s, p, and d shells for a total of 18 electrons in the 9 valence orbitals, he reasoned that metal complexes with 18 electrons might also exhibit particularly high stability. This "18 electron rule" (also called the effective atomic number rule) is analogous to the octet rule discussed in earlier courses and is essentially kinetic in origin. This is because a compound which makes full use of its valence orbitals has no low lying vacant orbitals into which electrons can be promoted to initiate thermal decomposition or into which donation can occur in the case of nucleophilic attack. We can understand the effectiveness of the 18 electron rule by considering the simple molecular orbital diagram of an octahedral complex ML6. The 18 electron rule is of limited use in ionic systems because the HOMO-LUMO gap is small (another way of stating this is to say that ∆o, from crystal field theory, is small) (Fig. 1a below). As the covalent character of the metal ligand * bonding increases, the size of the HOMO-LUMO gap also increases (because eg is higher in energy when the orbital overlap is better) (Fig. 1b, 1c and Fig. 2) and therefore the 18 electron rule becomes more important. Finally, in cases where the ligands can function as π-acceptors as well as σ-donors, the interaction of the t2g set of orbitals (remember these are σ-nonbonding) with the ligand π-symmetry orbitals results in an effective increase in the HOMO-LUMO gap (∆o in Fig. -

Application of Novel Phosphine Ligands in Palladium-Catalyzed Cross-Coupling Reactions
Leibniz-Institut für Katalyse e.V. an der Universität Rostock Application of Novel Phosphine Ligands in Palladium-Catalyzed Cross-Coupling Reactions Dissertation zur Erlangung des akademischen Grades doctor rerum naturalium (Dr. rer. nat.) der Mathematisch-Naturwissenschaftlichen Fakultät der Universität Rostock vorgelegt von Christian Torborg, geb am. 8.3.1980 in Stade Rostock, 3. April 2009 urn:nbn:de:gbv:28-diss2009-0174-3 ii Die vorliegende Arbeit entstand in der Zeit von April 2006 bis März 2009 am Leibniz- Institut für Katalyse e.V. an der Universität Rostock. Gutachter der Dissertation 1. Prof. Dr. Matthias Beller, Universität Rostock 2. Prof. Dr. Carsten Bolm, RWTH Aachen Termin der Rigorosumsprüfung: 11.6.2009 1. Prüfer: Prof. Dr. Matthias Beller (Hauptfach: Organische Chemie) 2. Prüfer: Prof. Dr. Friedemann W. Nerdinger (Nebenfach: Arbeits- und Organisationspsychologie) Termin der öffentlichen Verteidigung: 18.6.2009 iii Weil du nur einmal lebst willst du dass sich was bewegt bevor du gehst Du schreibst Geschichte an jedem Tag denn jetzt und hier bist du ein Teil von ihr Madsen, Du schreibst Geschichte iv Danksagung Mein besonderer Dank gebührt meinem Mentor Herrn Prof. Dr. Matthias Beller für die Aufnahme in die Arbeitsgruppe, die anregenden und motivierenden Diskussionen und den hervorragend ausgestatteten Arbeitsplatz. Darüberhinaus möchte ich meinen Themenleitern Dr. Alexander Zapf und Dr. Helfried Neumann für die unkomplizierte und freundliche Zusammenarbeit sowie die hilfreichen Disskussionen danken. Danken möchte ich vor allem auch meiner Arbeitsgruppe für die gute Zusammenarbeit: Dr. Anne Brennführer, Sandra Leiminger, Thomas ‚Schulle’ Schulz, Andreas ‚Dandy’ Dumrath, Dr. Alexey Sergeev, Dr. Pazhmalai Anbarasan und Dr. Thomas Schareina. Für ihre Geduld und die gute Arbeitsatmosphäre im Labor bin ich außerdem Dr. -

Inorganic Chemistry Ii
INORGANIC CHEMISTRY II OBJECTIVES 1. To understand the role of metal ions in biological process. 2. To learn the basic concepts of chemotherapy. 3. To learn the principle of catalysis and reaction mechanisms of organometallics. UNIT I: General Principles of Bioinorganic Chemistry Occurrence and availability of inorganic elements in biological systems – biomineralization – control and assembly of advanced materials in biology – nucleation and crystal growth – various biominerals – calcium phosphate – calcium carbonate – amorphous silica, iron biominerals – strontium and barium sulphate. Function and transport of alkali and alkaline earth metal ions: characterization of K+, Na+, Ca2+ and Mg2+ – complexes of alkali and alkaline earth metal ions with macrocycles – ion channels – ion pumps, catalysis and regulation of bioenergetic processes by the alkaline earth metal ions – Mg2+ and Ca2+. Metals at the center of photosynthesis – primary processes in photosynthesis – photosystems I and II-light absorption (energy acquisition) – exciton transport (direct energy transfer) – charge separation and electron transport – manganese catalyzed oxidation of water to O2. UNIT II: Amines, Proteins and Enzymes Cobalamines: reactions of the alkyl cobalamines – one electron reduction and oxidation – Co-C bond cleavage – coenzyme B12 – alkylation reactions of methylcobalamin. Heme and non-heme proteins – haemoglobin and myoglobin – oxygen transport and storage – electron transfer and oxygen activation – cytochromes, ferredoxins and rubredoxin – model systems, mononuclear non-heme iron enzymes. Copper containing proteins – classification and examples – electron transfer – oxygen transport-oxygenation – oxidases and reductases – cytochrome oxidase – superoxide dismutase (Cu, Zn) – nickel containing enzyme: urease. UNIT III: Medicinal Bioinorganic Chemistry Bioinorganic chemistry of quintessentially toxic metals – lead, cadmium, mercury, aluminium, chromium, copper and plutonium – detoxification by metal chelation – drugs that act by binding at the metal sites of metalloenzymes.