Synthesis and Structural Studies of Metallacarboranes Greig Scott
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

5.111 Principles of Chemical Science, Fall 2005 Transcript – Lecture 28
MIT OpenCourseWare http://ocw.mit.edu 5.111 Principles of Chemical Science, Fall 2005 Please use the following citation format: Sylvia Ceyer and Catherine Drennan, 5.111 Principles of Chemical Science, Fall 2005. (Massachusetts Institute of Technology: MIT OpenCourseWare). http://ocw.mit.edu (accessed MM DD, YYYY). License: Creative Commons Attribution-Noncommercial-Share Alike. Note: Please use the actual date you accessed this material in your citation. For more information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms MIT OpenCourseWare http://ocw.mit.edu 5.111 Principles of Chemical Science, Fall 2005 Transcript – Lecture 28 All right. We have a few more PowerPoint things before I am going to attempt using the board. Again, we are in the transition metal unit. And today we are going to introduce something called crystal field theory. And this is in Chapter 16 in your book. Let me just tell you about two different theories. Again, chemistry is an experimental science. We collect data and then we try to come up with theories that explain the data, so the theories are not sort of 100%. And some are more simple and some are more complicated to try to explain what we observe. And some of these theories, although they are pretty simple approximations of what is really going on, do a pretty good job of explaining the things that we are observing. All right. There are two different kinds of theories that you will hear about with transition metals. You will hear about crystal field theory and ligand field theory. -

Clusters – Contemporary Insight in Structure and Bonding 174 Structure and Bonding
Structure and Bonding 174 Series Editor: D.M.P. Mingos Stefanie Dehnen Editor Clusters – Contemporary Insight in Structure and Bonding 174 Structure and Bonding Series Editor: D.M.P. Mingos, Oxford, United Kingdom Editorial Board: X. Duan, Beijing, China L.H. Gade, Heidelberg, Germany Y. Lu, Urbana, IL, USA F. Neese, Mulheim€ an der Ruhr, Germany J.P. Pariente, Madrid, Spain S. Schneider, Gottingen,€ Germany D. Stalke, Go¨ttingen, Germany Aims and Scope Structure and Bonding is a publication which uniquely bridges the journal and book format. Organized into topical volumes, the series publishes in depth and critical reviews on all topics concerning structure and bonding. With over 50 years of history, the series has developed from covering theoretical methods for simple molecules to more complex systems. Topics addressed in the series now include the design and engineering of molecular solids such as molecular machines, surfaces, two dimensional materials, metal clusters and supramolecular species based either on complementary hydrogen bonding networks or metal coordination centers in metal-organic framework mate- rials (MOFs). Also of interest is the study of reaction coordinates of organometallic transformations and catalytic processes, and the electronic properties of metal ions involved in important biochemical enzymatic reactions. Volumes on physical and spectroscopic techniques used to provide insights into structural and bonding problems, as well as experimental studies associated with the development of bonding models, reactivity pathways and rates of chemical processes are also relevant for the series. Structure and Bonding is able to contribute to the challenges of communicating the enormous amount of data now produced in contemporary research by producing volumes which summarize important developments in selected areas of current interest and provide the conceptual framework necessary to use and interpret mega- databases. -

Organometrallic Chemistry
CHE 425: ORGANOMETALLIC CHEMISTRY SOURCE: OPEN ACCESS FROM INTERNET; Striver and Atkins Inorganic Chemistry Lecturer: Prof. O. G. Adeyemi ORGANOMETALLIC CHEMISTRY Definitions: Organometallic compounds are compounds that possess one or more metal-carbon bond. The bond must be “ionic or covalent, localized or delocalized between one or more carbon atoms of an organic group or molecule and a transition, lanthanide, actinide, or main group metal atom.” Organometallic chemistry is often described as a bridge between organic and inorganic chemistry. Organometallic compounds are very important in the chemical industry, as a number of them are used as industrial catalysts and as a route to synthesizing drugs that would not have been possible using purely organic synthetic routes. Coordinative unsaturation is a term used to describe a complex that has one or more open coordination sites where another ligand can be accommodated. Coordinative unsaturation is a very important concept in organotrasition metal chemistry. Hapticity of a ligand is the number of atoms that are directly bonded to the metal centre. Hapticity is denoted with a Greek letter η (eta) and the number of bonds a ligand has with a metal centre is indicated as a superscript, thus η1, η2, η3, ηn for hapticity 1, 2, 3, and n respectively. Bridging ligands are normally preceded by μ, with a subscript to indicate the number of metal centres it bridges, e.g. μ2–CO for a CO that bridges two metal centres. Ambidentate ligands are polydentate ligands that can coordinate to the metal centre through one or more atoms. – – – For example CN can coordinate via C or N; SCN via S or N; NO2 via N or N. -

Neutral All Metal Aromatic Half-Sandwich Complexes Between Alkaline Earth and Transition Metals: an Ab-Initio Exploration
Neutral All Metal Aromatic Half-Sandwich Complexes Between Alkaline Earth and Transition Metals: An Ab-initio Exploration Amlan Jyoti Kalita Cotton University Prem Prakash Sahu Cotton University Ritam Raj Borah Gauhati University Shahnaz Sultana Rohman Cotton University Chayanika Kashyap Cotton University Sabnam Swabaka Ullah Cotton University Indrani Baruah Cotton University Lakhya Jyoti Mazumder Cotton University Dimpul Konwar Gachon University Ankur Kanti Guha ( [email protected] ) Cotton University https://orcid.org/0000-0003-4370-8108 Research Article Keywords: Half-sandwich complexes, dual aromaticity, topological analyses Posted Date: May 24th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-505446/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/13 Version of Record: A version of this preprint was published at Structural Chemistry on July 7th, 2021. See the published version at https://doi.org/10.1007/s11224-021-01807-w. Page 2/13 Abstract Sandwich complexes nd their interests among the chemists after the breakthrough discovery of ferrocene. Since then, a number of sandwich and half sandwich complexes were predicted and synthesized. Herein, we have theoretically proposed a series of half-sandwich complexes involving a neutral Be3 ring and transition metal. Quantum chemical calculations have shown that the proposed complexes are quite stable involving high bond dissociation energies. The thermodynamics of their formation is also favorable. The Be3 ring in all cases posses dual aromaticity which has been ascertained based on magnetic as well as topological feature of electron density. Introduction Discovery of the rst sandwich complex (C5H5)2Fe, commonly known as “ferrocene”, drew the attention of the chemistry fraternity in no time [1–3]. -

A Ladder Polysilane As a Template for Folding Palladium Nanosheets
ARTICLE Received 15 Mar 2013 | Accepted 15 May 2013 | Published 14 Jun 2013 DOI: 10.1038/ncomms3014 A ladder polysilane as a template for folding palladium nanosheets Yusuke Sunada1,2, Ryohei Haige2, Kyohei Otsuka3, Soichiro Kyushin3 & Hideo Nagashima1,2 Although discrete nano-sized compounds consisting of a monolayer sheet of multiple atoms have attracted much attention, monolayer transition metal nanosheets are difficult to access. Here we report a template synthesis of the folding metal nanosheet (2) consisting of 11 palladium atoms by treatment of a ladder polysilane, decaisopropylbicyclo[2.2.0]hexasilane t (1), with Pd(CN Bu)2. Crystallographic analysis reveals that the compound is composed of two monolayer Pd7 sheets sharing three palladium atoms at the junction. Each Pd atom is stabilized by Pd–Si s-bonds, Pd–Pd bonds and coordination of isocyanides. Ligand exchange t of 2 from CN Bu to CN(2,4,6-Me3-C6H2) is accompanied by structural rearrangement, leading to the formation of another folding Pd11 nanosheet (3) consisting of two edge-sharing Pd7 sheets. The shapes of the Pd7 sheets as well as the dihedral angle between the two Pd7 sheets are dependent on the substituent of the isocyanide ligand. 1 Institute for Materials Chemistry and Engineering, Kyushu University and CREST, Japan Science and Technology Agency (JST), Kasuga, Fukuoka 816-8580, Japan. 2 Graduate School of Engineering Sciences, Kyushu University, 6-1 Kasugakoen, Kasuga, Fukuoka 816-8580, Japan. 3 Department of Chemistry and Chemical Biology, Graduate School of Engineering, Gunma University, Kiryu, Gunma 376-8515, Japan. Correspondence and requests for materials should be addressed to H.N. -

Organometallic and Catalysis
ORGANOMETALLIC AND CATALYSIS Dr. Malay Dolai, Assistant Professor, Department of Chemistry, Prabhat Kumar College, Contai, Purba Medinipur-721404, WB, India. 1.Introduction Organometallic chemistry is the study of organometallic compounds, chemical compounds containing at least one chemical bond between a carbon atom of an organic molecule and a metal, including alkaline, alkaline earth, and transition metals, and sometimes broadened to include metalloids like boron, silicon, and tin, as well. Aside from bonds to organyl fragments or molecules, bonds to 'inorganic' carbon, like carbon monoxide (metal carbonyls), cyanide, or carbide, are generally considered to be organometallic as well. Some related compounds such as transition metal hydrides and metal phosphine complexes are often included in discussions of organometallic compounds, though strictly speaking, they are not necessarily organometallic. The related but distinct term "metalorganic compound" refers to metal-containing compounds lacking direct metal-carbon bonds but which contain organic ligands. In 1827, Zeise's salt is the first platinum- olefin complex: K[PtCl3(C2H4)].H2O, the first invented organometallic compound. Organometallic compounds find wide use in commercial reactions, both as homogeneous catalysis and as stoichiometric reagents For instance, organolithium, organomagnesium, and organoaluminium compounds, examples of which are highly basic and highly reducing, are useful stoichiometrically, but also catalyze many polymerization reactions. Almost all processes involving carbon monoxide rely on catalysts, notable examples being described as carbonylations. The production of acetic acid from methanol and carbon monoxide is catalyzed via metal carbonyl complexes in the Monsanto process and Cativa process. Most synthetic aldehydes are produced via hydroformylation. The bulk of the synthetic alcohols, at least those larger than ethanol, are produced by hydrogenation of hydroformylation- derived aldehydes. -

Synthesis and Reactivity of Cyclopentadienyl Based Organometallic Compounds and Their Electrochemical and Biological Properties
Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties Sasmita Mishra Department of Chemistry National Institute of Technology Rourkela Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties Dissertation submitted to the National Institute of Technology Rourkela In partial fulfillment of the requirements of the degree of Doctor of Philosophy in Chemistry by Sasmita Mishra (Roll Number: 511CY604) Under the supervision of Prof. Saurav Chatterjee February, 2017 Department of Chemistry National Institute of Technology Rourkela Department of Chemistry National Institute of Technology Rourkela Certificate of Examination Roll Number: 511CY604 Name: Sasmita Mishra Title of Dissertation: ''Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties We the below signed, after checking the dissertation mentioned above and the official record book(s) of the student, hereby state our approval of the dissertation submitted in partial fulfillment of the requirements of the degree of Doctor of Philosophy in Chemistry at National Institute of Technology Rourkela. We are satisfied with the volume, quality, correctness, and originality of the work. --------------------------- Prof. Saurav Chatterjee Principal Supervisor --------------------------- --------------------------- Prof. A. Sahoo. Prof. G. Hota Member (DSC) Member (DSC) --------------------------- -

Metal-Ligand Bonding and Inorganic Reaction Mechanisms Year 2
Metal-Ligand Bonding and Inorganic Reaction Mechanisms Year 2 RED Metal-ligand and metal-metal bonding of the transition metal elements Synopsis Lecture 1: Trends of the transition metal series. Ionic vs Covalent bonding. Nomenclature. Electron counting. Lecture 2: Thermodynamics of complex formation. Why complexes form. Recap of molecular orbital theory. 18-electron rule. Lecture 3: Ligand classes. -donor complexes. Octahedral ML6 molecular orbital energy diagram. Lecture 3: - acceptor ligands and synergic bonding. Binding of CO, CN , N2, O2 and NO. Lecture 4: Alkenes, M(H2) vs M(H)2, Mn(O2) complexes, PR3. Lecture 5: 2- - 2- 3- donor ligands, metal-ligand multiple bonds, O , R2N , RN , N . Lecture 6: ML6 molecular orbital energy diagrams incorporating acceptor and donor ligands. Electron counting revisited and link to spectrochemical series. Lecture 7: Kinetics of complex formation. Substitution mechanisms of inorganic complexes. Isomerisation. Lecture 8: Ligand effects on substitution rates (trans-effect, trans-influence). Metal and geometry effects on substitution rates. Lecture 9: Outer sphere electron transfer. Lecture 10: Inner Sphere electron transfer. Bridging ligands. 2 Learning Objectives: by the end of the course you should be able to i) Use common nomenclature in transition metal chemistry. ii) Count valence electrons and determine metal oxidation state in transition metal complexes. iii) Understand the physical basis of the 18-electron rule. iv) Appreciate the synergic nature of bonding in metal carbonyl complexes. v) Understand the relationship between CO, the 'classic' -acceptor and related ligands such as NO, CN, N2, and alkenes. 2 vi) Describe the nature of the interaction between -bound diatomic molecules (H2, O2) and their relationship to -acceptor ligands. -

Materials Sciences Programs Fiscal Year 1994
DOE/ER-0648 Dist. Category UC-404 April 1995 Materials Sciences Programs Fiscal Year 1994 U.S. Department of Energy Office ofEnergy Research Office of Basic Energy Sciences Division of Materials Sciences Germantown Building 19901 Germantown Road Germantown, MD 20874-1290 FOREWORD The Division of Materials Sciences is located within the Department of Energy (DOE) in the Office of Basic Energy Sciences which is under the Office of Energy Research. The Director of the Office of Energy Research is appointed by the President and confirmed by the Senate. The Director of the Office of Energy Research is responsible for oversight of, and providing advice to, the Secretary of Energy on the Department's research portfolio and on the management of all of the Laboratories that It owns, except for those that are designated as having a primary role in nuclear weaponry. The Division of Materials Sciences Is responsible for basic research and research facilities in strategic materials science topics of critical importance to the mission of the Department and its Strategic Plan. Other programmatic divisions under the Office of Basic Energy Sciences are Chemical Sciences, Engineering and Geosciences, and Energy Biosciences; information for them is contained on page 165. Materials Science is an enabling technology. The performance parameters, economics, environmental acceptability and safety of all energy generation, conversion, transmission and conservation technologies are limited by the properties and behavior of materials. The Materials Sciences programs develop scientific understanding of the synergistic relationship amongst the synthesis, processing, structure, properties, behavior, performance and other characteristics of materials. Emphasis is placed on the development of the capability to discover technologically, economically, and environmentally desirable new materials and processes, and the instruments and national user facilities necessary for achieving such progress. -

Metal–Dithiolene Bonding Contributions to Pyranopterin Molybdenum Enzyme Reactivity
inorganics Review Metal–Dithiolene Bonding Contributions to Pyranopterin Molybdenum Enzyme Reactivity Jing Yang 1 , John H. Enemark 2 and Martin L. Kirk 1,* 1 Department of Chemistry and Chemical Biology, The University of New Mexico, MSC03 2060, Albuquerque, NM 87131-0001, USA; [email protected] 2 Department of Chemistry Biochemistry, University of Arizona, Tucson, AZ 85721, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-505-277-5992 Received: 2 February 2020; Accepted: 2 March 2020; Published: 5 March 2020 Abstract: Here we highlight past work on metal–dithiolene interactions and how the unique electronic structure of the metal–dithiolene unit contributes to both the oxidative and reductive half reactions in pyranopterin molybdenum and tungsten enzymes. The metallodithiolene electronic structures detailed here were interrogated using multiple ground and excited state spectroscopic probes on the enzymes and their small molecule analogs. The spectroscopic results have been interpreted in the context of bonding and spectroscopic calculations, and the pseudo-Jahn–Teller effect. The dithiolene is a unique ligand with respect to its redox active nature, electronic synergy with the pyranopterin component of the molybdenum cofactor, and the ability to undergo chelate ring distortions that control covalency, reduction potential, and reactivity in pyranopterin molybdenum and tungsten enzymes. Keywords: metal–dithiolene; pyranopterin molybdenum enzymes; fold-angle; tungsten enzymes; electronic structure; pseudo-Jahn–Teller effect; thione; molybdenum cofactor; Moco 1. Introduction It is now well-established that all known molybdenum-containing enzymes [1–3], with the sole exception of nitrogenase, contain a common pyranopterin dithiolene (PDT) (Figure1) organic cofactor (originally called molybdopterin (MPT)), which coordinates to the Mo center of the enzymes through the sulfur atoms of the dithiolene fragment. -

21.1-21.4, 22 Ch
Assigned Reading: 21.1-21.4, 22 Ch 102 – Problem Set 6 Due : Thursday, May 20 – Before Class Problem 1 (20 pts) For the following molecules, (i) Determine the oxidation state and d electron count of the metal (ii) Identify the compound as high spin or low spin (iii) Calculate the spin-only magnetic moment based on the number of unpaired electrons 3- a) [FeBr6] 2+ b) [Co(NH3)6] 3+ c) [Co(NH3)6] 3+ d) [Mn(CO)6] 3- e) [OsBr6] 2- f) [RhF6] g) TiCl4 - h) [TiCl4] Problem 2 (20 pts) For the following transition metal complexes, propose the most reasonable structure. Keep in mind the 18-electron rule. Give the valence shell electron count, the formal oxidation state and the d-electron count. Make sure that the ligand hapticity is clear in your drawings. Indicate clearly whether the NO ligand is bent or linear, if applicable. a. (C6H6)Fe(CO)2PPh3 2+ b. [(C6H6)Fe(CO)2PPh3] c. Fe(NO)2(CO)2 d. (C6Me6)2Ru (C6Me6 = hexamethylbenzene) e. Mn2(CO)10 f. [(C6H6)W(C3H5)]2(µ-Cl)2 + g. [CoCl(en)2(NO)] (en = ethane-1,2-diamine, a.k.a. ethylenediamine) Problem 3 (15 Points) For the following transition metal complexes, give the valence shell electron count, the formal oxidation state, and the d-electron count. a. PR2 Ph B Fe N R = H2C P R 2PR2 b. tBuO OtBu OtBu W W tBuO tBuO OtBu c. (H3C)2HC Cl Cl CH(CH3)2 (H C) HC CH(CH ) 3 2 P Ni Ni P 3 2 d. -
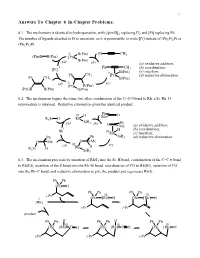
Answers to Chapter 6 In-Chapter Problems
1 Answers To Chapter 6 In-Chapter Problems. 6.1. The mechanism is identical to hydrogenation, with [(pin)B]2 replacing H2 and [Pt] replacing Pd. The number of ligands attached to Pt is uncertain, so it is permissible to write [Pt] instead of (Ph3P)2Pt or (Ph3P)3Pt. II B(Pin) Ph CH3 (Pin)B B(Pin) [Pt] B(Pin) (a) (b) (a) oxidative addition; 0 Ph CH (b) coordination; [Pt] 3 II B(Pin) (c) insertion; Ph CH3 [Pt] (d) reductive elimination. Ph CH3 B(Pin) (d) II (c) [Pt] B(Pin) (Pin)B B(Pin) B(Pin) 6.2. The mechanism begins the same, but after coordination of the C=O π bond to Rh, a Si–Rh–O intermediate is obtained. Reductive elimination gives the identical product. Ph III H Me O R3Si H Rh (a) SiR3 (b) Ph O Me (a) oxidative addition; I H (b) coordination; Rh IIIRh (c) insertion; Ph Ph SiR3 (d) reductive elimination. O Me O Me III (c) (d) Rh H R3Si H SiR3 6.3. The mechanism proceeds by insertion of Rh(I) into the Si–H bond, coordination of the C=C π bond to Rh(III), insertion of the π bond into the Rh–Si bond, coordination of CO to Rh(III), insertion of CO into the Rh–C bond, and reductive elimination to give the product and regenerate Rh(I). Ph Ph OSi H Ph Ph Ph Ph i-Pr III III I OSi [Rh] H OSi [Rh] H [Rh] i-Pr i-Pr product Ph Ph H Ph Ph H Ph Ph III III III OSi [Rh] C O OSi [Rh] C O OSi [Rh] H i-Pr i-Pr i-Pr Chapter 6 2 6.4.