High Expression of PU.1 Is Associated with Her‑2 and Shorter Survival in Patients with Breast Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of PU.1 and GATA-1 Transcription Factors During Normal and Leukemogenic Hematopoiesis
Leukemia (2010) 24, 1249–1257 & 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 www.nature.com/leu REVIEW The role of PU.1 and GATA-1 transcription factors during normal and leukemogenic hematopoiesis P Burda1, P Laslo2 and T Stopka1,3 1Department of Pathophysiology and Center of Experimental Hematology, First Faculty of Medicine, Charles University, Prague, Czech Republic; 2Section of Experimental Haematology, Leeds Institute of Molecular Medicine, University of Leads, St James’s University Hospital, Leeds, UK and 31st Department of Medicine-Hematology, General University Hospital, Prague, Czech Republic Hematopoiesis is coordinated by a complex regulatory network Additional domains include an N-terminal acidic domain and a of transcription factors and among them PU.1 (Spi1, Sfpi1) glutamine-rich domain, both involved in transcriptional activa- represents a key molecule. This review summarizes the tion, as well as a PEST domain involved in protein–protein indispensable requirement of PU.1 during hematopoietic cell fate decisions and how the function of PU.1 can be modulated interactions. PU.1 protein can be modified post-translationally by protein–protein interactions with additional factors. The by phosporylation at serines 41 (N-terminal acidic domain) and mutual negative regulation between PU.1 and GATA-1 is 142 and 148 (PEST domain), which results in augmented detailed within the context of normal and leukemogenic activity. hematopoiesis and the concept of ‘differentiation therapy’ to The PU.1 protein can physically interact with a variety of restore normal cellular differentiation of leukemic cells is regulatory factors including (i) general transcription factors discussed. Leukemia (2010) 24, 1249–1257; doi:10.1038/leu.2010.104; (TFIID, TBP), (ii) early hematopoietic transcription factors published online 3 June 2010 (GATA-2 and Runx-1), (iii) erythroid factor (GATA-1) and (iv) Keywords: PU.1; leukemia differentiation; GATA-1; chromatin; non-erythroid factors (C/EBPa, C/EBPb, IRF4/8 and c-Jun). -

Activated Peripheral-Blood-Derived Mononuclear Cells
Transcription factor expression in lipopolysaccharide- activated peripheral-blood-derived mononuclear cells Jared C. Roach*†, Kelly D. Smith*‡, Katie L. Strobe*, Stephanie M. Nissen*, Christian D. Haudenschild§, Daixing Zhou§, Thomas J. Vasicek¶, G. A. Heldʈ, Gustavo A. Stolovitzkyʈ, Leroy E. Hood*†, and Alan Aderem* *Institute for Systems Biology, 1441 North 34th Street, Seattle, WA 98103; ‡Department of Pathology, University of Washington, Seattle, WA 98195; §Illumina, 25861 Industrial Boulevard, Hayward, CA 94545; ¶Medtronic, 710 Medtronic Parkway, Minneapolis, MN 55432; and ʈIBM Computational Biology Center, P.O. Box 218, Yorktown Heights, NY 10598 Contributed by Leroy E. Hood, August 21, 2007 (sent for review January 7, 2007) Transcription factors play a key role in integrating and modulating system. In this model system, we activated peripheral-blood-derived biological information. In this study, we comprehensively measured mononuclear cells, which can be loosely termed ‘‘macrophages,’’ the changing abundances of mRNAs over a time course of activation with lipopolysaccharide (LPS). We focused on the precise mea- of human peripheral-blood-derived mononuclear cells (‘‘macro- surement of mRNA concentrations. There is currently no high- phages’’) with lipopolysaccharide. Global and dynamic analysis of throughput technology that can precisely and sensitively measure all transcription factors in response to a physiological stimulus has yet to mRNAs in a system, although such technologies are likely to be be achieved in a human system, and our efforts significantly available in the near future. To demonstrate the potential utility of advanced this goal. We used multiple global high-throughput tech- such technologies, and to motivate their development and encour- nologies for measuring mRNA levels, including massively parallel age their use, we produced data from a combination of two distinct signature sequencing and GeneChip microarrays. -

The Unique Transcriptional Response Produced by Concurrent Estrogen
Need et al. BMC Cancer (2015) 15:791 DOI 10.1186/s12885-015-1819-3 RESEARCH ARTICLE Open Access The unique transcriptional response produced by concurrent estrogen and progesterone treatment in breast cancer cells results in upregulation of growth factor pathways and switching from a Luminal A to a Basal-like subtype Eleanor F. Need1*,LukeA.Selth2,3,AndrewP.Trotta1,4,DamienA.Leach1,LaurenGiorgio1, Melissa A. O’Loughlin1, Eric Smith5, Peter G. Gill6,WendyV.Ingman7,8, J. Dinny Graham9 and Grant Buchanan1,3 Abstract Background: In breast cancer, progesterone receptor (PR) positivity or abundance is positively associated with survival and treatment response. It was initially believed that PR was a useful diagnostic marker of estrogen receptor activity, but increasingly PR has been recognised to play an important biological role in breast homeostasis, carcinogenesis and metastasis. Although PR expression is almost exclusively observed in estrogen receptor positive tumors, few studies have investigated the cellular mechanisms of PR action in the context of ongoing estrogen signalling. Methods: In this study, we contrast PR function in estrogen pretreated ZR-75-1 breast cancer cells with vehicle treated ZR-75-1 and T-47D breast cancer cells using expression microarrays and chromatin immunoprecipitation-sequencing. Results: Estrogen cotreatment caused a dramatic increase in the number of genes regulated by progesterone in ZR-75-1 cells. In T-47D cells that have naturally high levels of PR, estrogen and progesterone cotreatment resulted in a reduction in the number of regulated genes in comparison to treatment with either hormone alone. At a genome level, estrogen pretreatment of ZR-75-1 cells led to a 10-fold increase in the number of PR DNA binding sites detected using ChIP-sequencing. -

HSF1) During Macrophage Differentiation of Monocytes
Leukemia (2014) 28, 1676–1686 & 2014 Macmillan Publishers Limited All rights reserved 0887-6924/14 www.nature.com/leu ORIGINAL ARTICLE Dual regulation of SPI1/PU.1 transcription factor by heat shock factor 1 (HSF1) during macrophage differentiation of monocytes G Jego1,2, D Lanneau1,2, A De Thonel1,2, K Berthenet1,2, A Hazoume´ 1,2, N Droin3,4, A Hamman1,2, F Girodon1,2, P-S Bellaye1,2, G Wettstein1,2, A Jacquel1,2,5, L Duplomb6,7, A Le Moue¨l8,9, C Papanayotou10, E Christians11, P Bonniaud1,2, V Lallemand-Mezger8,9, E Solary3,4 and C Garrido1,2,12 In addition to their cytoprotective role in stressful conditions, heat shock proteins (HSPs) are involved in specific differentiation pathways, for example, we have identified a role for HSP90 in macrophage differentiation of human peripheral blood monocytes that are exposed to macrophage colony-stimulating factor (M-CSF). Here, we show that deletion of the main transcription factor involved in heat shock gene regulation, heat shock factor 1 (HSF1), affects M-CSF-driven differentiation of mouse bone marrow cells. HSF1 transiently accumulates in the nucleus of human monocytes undergoing macrophage differentiation, including M-CSF- treated peripheral blood monocytes and phorbol ester-treated THP1 cells. We demonstrate that HSF1 has a dual effect on SPI1/ PU.1, a transcription factor essential for macrophage differentiation and whose deregulation can lead to the development of leukemias and lymphomas. Firstly, HSF1 regulates SPI1/PU.1 gene expression through its binding to a heat shock element within the intron 2 of this gene. -
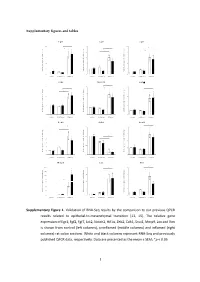
Type of the Paper (Article
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1. -

Functional Annotation of Human Long Noncoding Rnas Using Chromatin
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.13.426305; this version posted January 14, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Funconal annotaon of human long noncoding RNAs using chroman conformaon data Saumya Agrawal1, Tanvir Alam2, Masaru Koido1,3, Ivan V. Kulakovskiy4,5, Jessica Severin1, Imad ABugessaisa1, Andrey Buyan5,6, Josee Dos&e7, Masayoshi Itoh1,8, Naoto Kondo9, Yunjing Li10, Mickaël Mendez11, Jordan A. Ramilowski1,12, Ken Yagi1, Kayoko Yasuzawa1, CHi Wai Yip1, Yasushi Okazaki1, MicHael M. Ho9man11,13,14,15, Lisa Strug10, CHung CHau Hon1, CHikashi Terao1, Takeya Kasukawa1, Vsevolod J. Makeev4,16, Jay W. SHin1, Piero Carninci1, MicHiel JL de Hoon1 1RIKEN Center for Integra&ve Medical Sciences, YokoHama, Japan. 2College of Science and Engineering, Hamad Bin KHalifa University, DoHa, Qatar. 3Ins&tute of Medical Science, THe University of Tokyo, Tokyo, Japan. 4Vavilov Ins&tute of General Gene&cs, Russian Academy of Sciences, Moscow, Russia. 5Ins&tute of Protein ResearcH, Russian Academy of Sciences, PusHcHino, Russia. 6Faculty of Bioengineering and Bioinforma&cs, Lomonosov Moscow State University, Moscow, Russia. 7Department of BiocHemistry, Rosalind and Morris Goodman Cancer ResearcH Center, McGill University, Montréal, QuéBec, Canada. 8RIKEN Preven&ve Medicine and Diagnosis Innova&on Program, Wako, Japan. 9RIKEN Center for Life Science TecHnologies, YokoHama, Japan. 10Division of Biosta&s&cs, Dalla Lana ScHool of PuBlic HealtH, University of Toronto, Toronto, Ontario, Canada. 11Department of Computer Science, University of Toronto, Toronto, Ontario, Canada. 12Advanced Medical ResearcH Center, YokoHama City University, YokoHama, Japan. -

Logical Modeling of Lymphoid and Myeloid Cell Specification and Transdifferentiation
Logical modeling of lymphoid and myeloid cell specification and transdifferentiation Samuel Collombeta,1, Chris van Oevelenb,2, Jose Luis Sardina Ortegab,2, Wassim Abou-Jaoudéa, Bruno Di Stefanob,3, Morgane Thomas-Cholliera, Thomas Grafb,c,1, and Denis Thieffrya,1 aComputational Systems Biology Team, Institut de Biologie de l’Ecole Normale Supérieure, CNRS UMR8197, INSERM U1024, Ecole Normale Supérieure, Paris Sciences et Lettres Research University, 75005 Paris, France; bHematopoietic Stem Cells, Transdifferentiation, and Reprogramming Team, Gene Regulation, Stem Cells, and Cancer Program, Center for Genomic Regulation, Barcelona Institute for Biotechnology, 08003 Barcelona, Spain; and cUniversitat Pompeu Fabra, 08002 Barcelona, Spain Edited by Ellen V. Rothenberg, California Institute of Technology, Pasadena, CA, and accepted by Editorial Board Member Neil H. Shubin November 18, 2016 (received for review September 1, 2016) Blood cells are derived from a common set of hematopoietic stem for the transition from common myeloid progenitors (CMPs) to cells, which differentiate into more specific progenitors of the myeloid granulocyte-macrophage progenitors (GMPs), and mutation in and lymphoid lineages, ultimately leadingtodifferentiatedcells.This this gene can result in acute myeloid leukemia (6). Understanding developmental process is controlled by a complex regulatory network the molecular mechanisms by which such factors can induce cell- involving cytokines and their receptors, transcription factors, and fate decisions is of primary importance and might help in the chromatin remodelers. Using public data and data from our own mo- development of novel therapeutic strategies. lecular genetic experiments (quantitative PCR, Western blot, EMSA) or Computational modeling of regulatory networks is increasingly genome-wide assays (RNA-sequencing, ChIP-sequencing), we have recognized as a valuable approach to study cell-fate decisions. -

For Personal Use. Only Reproduce with Permission from the Lancet
Correlation to non-HGNT Accesion group 2 Description AA897204 1.479917 ESTs T85111 1.286576 null T60168 | AI821353 1.274487 thyroid transcription factor 1 AA600173 1.183065 ubiquitin-conjugating enzyme E2A (RAD6 homolog) R55745 1.169339 ELAV (embryonic lethal, abnormal vision, Drosophila)-like 4 (Hu antigen D) AA400492 1.114935 ESTs AA864791 1.088826 hypothetical protein FLJ21313 N53758 1.070402 EST AI216628 1.067763 ESTs AI167637 1.058561 ESTs AA478265 1.056331 similar to transmembrane receptor Unc5H1 AA969924 1.039315 EST AI074650 1.039043 hypothetical protein FLJ13842 R20763 1.035807 ELAV (embryonic lethal, abnormal vision, Drosophila)-like 3 (Hu antigen C) AI347081 1.034518 Ca2+-dependent activator protein for secretion R44386 1.028005 ESTs AA976699 1.027227 chromogranin A (parathyroid secretory protein 1) AA634475 1.026766 KIAA1796 protein AA496809 1.02432 SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 1 H16572 1.013059 null H29013 1.002117 seizure related 6 homolog (mouse)-like AI299731 1.001053 Homo sapiens nanos mRNA, partial cds AA400194 0.9950039 EST AI216537 | AI820870 0.9737153 Homo sapiens cDNA FLJ39674 fis, clone SMINT2009505 AA426408 0.9728649 type I transmembrane receptor (seizure-related protein) AA971742 | AI733380 0.9707561 achaete-scute complex-like 1 (Drosophila) R41450 0.9655133 ESTs AA487505 0.9636143 immunoglobulin superfamily, member 4 AA404337 0.957686 thymus high mobility group box protein TOX N68578 0.9552571 ESTs R45008 0.9422938 ELAV (embryonic lethal, abnormal -

The MER41 Family of Hervs Is Uniquely Involved in the Immune-Mediated Regulation of Cognition/Behavior-Related Genes
bioRxiv preprint doi: https://doi.org/10.1101/434209; this version posted October 3, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The MER41 family of HERVs is uniquely involved in the immune-mediated regulation of cognition/behavior-related genes: pathophysiological implications for autism spectrum disorders Serge Nataf*1, 2, 3, Juan Uriagereka4 and Antonio Benitez-Burraco 5 1CarMeN Laboratory, INSERM U1060, INRA U1397, INSA de Lyon, Lyon-Sud Faculty of Medicine, University of Lyon, Pierre-Bénite, France. 2 University of Lyon 1, Lyon, France. 3Banque de Tissus et de Cellules des Hospices Civils de Lyon, Hôpital Edouard Herriot, Lyon, France. 4Department of Linguistics and School of Languages, Literatures & Cultures, University of Maryland, College Park, USA. 5Department of Spanish, Linguistics, and Theory of Literature (Linguistics). Faculty of Philology. University of Seville, Seville, Spain * Corresponding author: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/434209; this version posted October 3, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. ABSTRACT Interferon-gamma (IFNa prototypical T lymphocyte-derived pro-inflammatory cytokine, was recently shown to shape social behavior and neuronal connectivity in rodents. STAT1 (Signal Transducer And Activator Of Transcription 1) is a transcription factor (TF) crucially involved in the IFN pathway. -

Epstein–Barr Virus Nuclear Antigen 3C Binds to BATF/IRF4 Or SPI1/IRF4 Composite Sites and Recruits Sin3a to Repress CDKN2A
Epstein–Barr Virus Nuclear Antigen 3C binds to BATF/IRF4 or SPI1/IRF4 composite sites and recruits Sin3A to repress CDKN2A Sizun Jianga,b,c,d, Bradford Willoxa,c, Hufeng Zhoua,c, Amy M. Holthausa,c, Anqi Wange, Tommy T. Shia,c, Seiji Maruof, Peter V. Kharchenkod,g, Eric C. Johannsene, Elliott Kieffa,b,c,1, and Bo Zhaoa,c aDepartment of Medicine, Brigham and Women’s Hospital, bProgram in Virology, cDepartment of Microbiology and Immunobiology, and dCenter for Biomedical Informatics, Harvard Medical School, Boston, MA 02115; eDepartment of Medicine and McArdle Laboratory for Cancer Research, University of Wisconsin–Madison, Madison, WI 53706; fDepartment of Tumor Virology, Institute for Genetic Medicine, Hokkaido University, Sapporo 060-0815, Japan; and gDivision of Hematology/Oncology, Children’s Hospital, Boston, MA 02115 Contributed by Elliott Kieff, November 20, 2013 (sent for review October 3, 2013) Epstein–Barr virus nuclear antigen 3C (EBNA3C) repression of LCL growth, indicating that EBNA3C is specifically required for CDKN2A p14ARF and p16INK4A is essential for immortal human LCL growth (6). Similar experiments reveal EBNA3C N-termi- B-lymphoblastoid cell line (LCL) growth. EBNA3C ChIP-sequencing nal amino acids 50–400 to be essential for LCL growth (3, 7, 8). identified >13,000 EBNA3C sites in LCL DNA. Most EBNA3C sites EBNA3C also up-regulates EBV LMP1 and cell CXCR4 and were associated with active transcription; 64% were strong CXCL12 gene expression (9–12), which are required for LCL H3K4me1- and H3K27ac-marked enhancers and 16% were active growth in nude mice (13). EBNA3C and EBNA3A joint re- p14ARF p16INK4A promoters marked by H3K4me3 and H3K9ac. -

An Interferon Signature Identified by RNA-Sequencing of Mammary Tissues Varies Across the Estrous Cycle and Is Predictive of Metastasis-Free Survival
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 5, No. 12 An interferon signature identified by RNA-sequencing of mammary tissues varies across the estrous cycle and is predictive of metastasis-free survival Antoine M. Snijders1, Sasha Langley1, Jian-Hua Mao1, Sandhya Bhatnagar1, Kathleen A. Bjornstad1, Chris J. Rosen1, Alvin Lo1, Yurong Huang1, Eleanor A. Blakely1, Gary H. Karpen1, Mina J. Bissell1 and Andrew J. Wyrobek1 1 Life Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA Correspondence to: Antoine M. Snijders, email: [email protected] Keywords: estrous cycle, mammary gland, RNA-sequencing, Type-1 interferon, low-dose ionizing radiation (LDIR), immunity, breast cancer, genetic susceptibility Received: May 31, 2014 Accepted: June 28, 2014 Published: June 30, 2014 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT The concept that a breast cancer patient’s menstrual stage at the time of tumor surgery influences risk of metastases remains controversial. The scarcity of comprehensive molecular studies of menstrual stage-dependent fluctuations in the breast provides little insight. To gain a deeper understanding of the biological changes in mammary tissue and blood during the menstrual cycle and to determine the influence of environmental exposures, such as low-dose ionizing radiation (LDIR), we used the mouse to characterize estrous-cycle variations in mammary gene transcripts by RNA-sequencing, peripheral white blood cell (WBC) counts and plasma cytokine levels. We identified an estrous-variable and hormone-dependent gene cluster enriched for Type-1 interferon genes. -

Micro RNA-Based Regulation of Genomics and Transcriptomics of Inflammatory Cytokines in COVID-19
medRxiv preprint doi: https://doi.org/10.1101/2021.06.08.21258565; this version posted June 12, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Micro RNA-based regulation of genomics and transcriptomics of inflammatory cytokines in COVID-19 Manoj Khokhar1, Sojit Tomo1, Purvi Purohit*1 Department of Biochemistry, All India Institute of Medical Sciences, Jodhpur 342005, India *Corresponding author and address Dr Purvi Purohit Additional Professor Department of Biochemistry All India Institute of Medical Sciences, Basni Industrial Area, Phase-2 Jodhpur-342005, India. Tel: 09928388223 NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2021.06.08.21258565; this version posted June 12, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Abstract: Background: Coronavirus disease 2019 is characterized by the elevation of a wide spectrum of inflammatory mediators, which are associated with poor disease outcomes. We aimed at an in-silico analysis of regulatory microRNA and their transcription factors (TF) for these inflammatory genes that may help to devise potential therapeutic strategies in the future.