Supplementary Figure 1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

A Genome-Wide Association Study Identifies New Susceptibility Loci for Esophageal Adenocarcinoma and Barrett's Esophagus
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by White Rose Research Online This is an author produced version of A genome-wide association study identifies new susceptibility loci for esophageal adenocarcinoma and Barrett's esophagus.. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/105073/ Article: Levine, D.M., Ek, W.E., Zhang, R. et al. (31 more authors) (2013) A genome-wide association study identifies new susceptibility loci for esophageal adenocarcinoma and Barrett's esophagus. Nature Genetics, 45 (12). pp. 1487-1493. ISSN 1061-4036 https://doi.org/10.1038/ng.2796 promoting access to White Rose research papers [email protected] http://eprints.whiterose.ac.uk/ HHS Public Access Author manuscript Author Manuscript Author ManuscriptNat Genet Author Manuscript. Author manuscript; Author Manuscript available in PMC 2014 June 01. Published in final edited form as: Nat Genet. 2013 December ; 45(12): 1487–1493. doi:10.1038/ng.2796. A Genome-Wide Association Study Identifies New Susceptibility Loci for Esophageal Adenocarcinoma and Barrett’ s Esophagus David M. Levine1, Weronica E. Ek2, Rui Zhang1, Xinxue Liu3, Lynn Onstad4, Cassandra Sather5, Pierre Lao-Sirieix3, Marilie D. Gammon6, Douglas A. Corley7, Nicholas J. Shaheen8, Nigel C. Bird9, Laura J. Hardie10, Liam J. Murray11, Brian J. Reid4,12, Wong-Ho Chow13, Harvey A. Risch14, Olof Nyrén15, Weimin Ye15, Geoffrey Liu16, Yvonne Romero17,18, Leslie Bernstein19, Anna H. Wu20, Alan G. Casson21, Stephen Chanock22, Patricia Harrington23,24,25, Isabel Caldas25, Irene Debiram-Beecham3, Carlos Caldas25,26, Nicholas K. Hayward27, Paul Pharoah23,24,25, Rebecca Fitzgerald3, Stuart MacGregor2, David C. -

Symplekin and Transforming Acidic Coiled-Coil Containing Protein 3 Support the Cancer Cell Mitotic Spindle
SYMPLEKIN AND TRANSFORMING ACIDIC COILED-COIL CONTAINING PROTEIN 3 SUPPORT THE CANCER CELL MITOTIC SPINDLE Kathryn M. Cappell A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctorate of Philosophy in the Department of Pharmacology, School of Medicine. Chapel Hill 2011 Approved by: Advisor: Dr. Angelique Whitehurst Reader: Dr. David Siderovski Reader: Dr. Channing Der Reader: Dr. Pilar Blancafort Reader: Dr. Mohanish Deshmukh ABSTRACT KATHRYN CAPPELL: Symplekin and Transforming Acidic Coiled-Coil Containing Protein 3 Support the Cancer Cell Mitotic Spindle (Under the direction of Dr. Angelique Whitehurst) An increased rate of proliferation in cancer cells, combined with abnormalities in spindle architecture, places tumors under increased mitotic stress. Previously, our laboratory performed a genome-wide paclitaxel chemosensitizer screen to identify genes whose depletion sensitizes non- small cell lung cancer (NSCLC) cells to mitotic stress induced by paclitaxel treatment. This screen uncovered a cohort of genes that are required for viability only in the presence of paclitaxel. Two genes uncovered in this screen were the polyadenylation scaffold symplekin and the gametogenic protein transforming acidic coiled-coil containing protein 3 (TACC3). Herein, we examine the impact of polyadenylation and gametogenesis on the tumor cell mitotic spindle. First, we demonstrate that depletion of SYMPK and other polyadenylation components sensitizes many NSCLC cells, but not normal immortalized lines, to paclitaxel by inducing mitotic errors and leading to abnormal mitotic progression. Second, we demonstrate that multiple gametogenic genes are required for normal microtubule dynamics and mitotic spindle formation in the presence of paclitaxel. -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

The Transformation of the Centrosome Into the Basal Body: Similarities and Dissimilarities Between Somatic and Male Germ Cells and Their Relevance for Male Fertility
cells Review The Transformation of the Centrosome into the Basal Body: Similarities and Dissimilarities between Somatic and Male Germ Cells and Their Relevance for Male Fertility Constanza Tapia Contreras and Sigrid Hoyer-Fender * Göttingen Center of Molecular Biosciences, Johann-Friedrich-Blumenbach Institute for Zoology and Anthropology-Developmental Biology, Faculty of Biology and Psychology, Georg-August University of Göttingen, 37077 Göttingen, Germany; [email protected] * Correspondence: [email protected] Abstract: The sperm flagellum is essential for the transport of the genetic material toward the oocyte and thus the transmission of the genetic information to the next generation. During the haploid phase of spermatogenesis, i.e., spermiogenesis, a morphological and molecular restructuring of the male germ cell, the round spermatid, takes place that includes the silencing and compaction of the nucleus, the formation of the acrosomal vesicle from the Golgi apparatus, the formation of the sperm tail, and, finally, the shedding of excessive cytoplasm. Sperm tail formation starts in the round spermatid stage when the pair of centrioles moves toward the posterior pole of the nucleus. The sperm tail, eventually, becomes located opposed to the acrosomal vesicle, which develops at the anterior pole of the nucleus. The centriole pair tightly attaches to the nucleus, forming a nuclear membrane indentation. An Citation: Tapia Contreras, C.; articular structure is formed around the centriole pair known as the connecting piece, situated in the Hoyer-Fender, S. The Transformation neck region and linking the sperm head to the tail, also named the head-to-tail coupling apparatus or, of the Centrosome into the Basal in short, HTCA. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

The Kinesin Spindle Protein Inhibitor Filanesib Enhances the Activity of Pomalidomide and Dexamethasone in Multiple Myeloma
Plasma Cell Disorders SUPPLEMENTARY APPENDIX The kinesin spindle protein inhibitor filanesib enhances the activity of pomalidomide and dexamethasone in multiple myeloma Susana Hernández-García, 1 Laura San-Segundo, 1 Lorena González-Méndez, 1 Luis A. Corchete, 1 Irena Misiewicz- Krzeminska, 1,2 Montserrat Martín-Sánchez, 1 Ana-Alicia López-Iglesias, 1 Esperanza Macarena Algarín, 1 Pedro Mogollón, 1 Andrea Díaz-Tejedor, 1 Teresa Paíno, 1 Brian Tunquist, 3 María-Victoria Mateos, 1 Norma C Gutiérrez, 1 Elena Díaz- Rodriguez, 1 Mercedes Garayoa 1* and Enrique M Ocio 1* 1Centro Investigación del Cáncer-IBMCC (CSIC-USAL) and Hospital Universitario-IBSAL, Salamanca, Spain; 2National Medicines Insti - tute, Warsaw, Poland and 3Array BioPharma, Boulder, Colorado, USA *MG and EMO contributed equally to this work ©2017 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2017.168666 Received: March 13, 2017. Accepted: August 29, 2017. Pre-published: August 31, 2017. Correspondence: [email protected] MATERIAL AND METHODS Reagents and drugs. Filanesib (F) was provided by Array BioPharma Inc. (Boulder, CO, USA). Thalidomide (T), lenalidomide (L) and pomalidomide (P) were purchased from Selleckchem (Houston, TX, USA), dexamethasone (D) from Sigma-Aldrich (St Louis, MO, USA) and bortezomib from LC Laboratories (Woburn, MA, USA). Generic chemicals were acquired from Sigma Chemical Co., Roche Biochemicals (Mannheim, Germany), Merck & Co., Inc. (Darmstadt, Germany). MM cell lines, patient samples and cultures. Origin, authentication and in vitro growth conditions of human MM cell lines have already been characterized (17, 18). The study of drug activity in the presence of IL-6, IGF-1 or in co-culture with primary bone marrow mesenchymal stromal cells (BMSCs) or the human mesenchymal stromal cell line (hMSC–TERT) was performed as described previously (19, 20). -

Identification of Genomic Targets of Krüppel-Like Factor 9 in Mouse Hippocampal
Identification of Genomic Targets of Krüppel-like Factor 9 in Mouse Hippocampal Neurons: Evidence for a role in modulating peripheral circadian clocks by Joseph R. Knoedler A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Neuroscience) in the University of Michigan 2016 Doctoral Committee: Professor Robert J. Denver, Chair Professor Daniel Goldman Professor Diane Robins Professor Audrey Seasholtz Associate Professor Bing Ye ©Joseph R. Knoedler All Rights Reserved 2016 To my parents, who never once questioned my decision to become the other kind of doctor, And to Lucy, who has pushed me to be a better person from day one. ii Acknowledgements I have a huge number of people to thank for having made it to this point, so in no particular order: -I would like to thank my adviser, Dr. Robert J. Denver, for his guidance, encouragement, and patience over the last seven years; his mentorship has been indispensable for my growth as a scientist -I would also like to thank my committee members, Drs. Audrey Seasholtz, Dan Goldman, Diane Robins and Bing Ye, for their constructive feedback and their willingness to meet in a frequently cold, windowless room across campus from where they work -I am hugely indebted to Pia Bagamasbad and Yasuhiro Kyono for teaching me almost everything I know about molecular biology and bioinformatics, and to Arasakumar Subramani for his tireless work during the home stretch to my dissertation -I am grateful for the Neuroscience Program leadership and staff, in particular -

A SARS-Cov-2 Protein Interaction Map Reveals Targets for Drug Repurposing
Article A SARS-CoV-2 protein interaction map reveals targets for drug repurposing https://doi.org/10.1038/s41586-020-2286-9 A list of authors and affiliations appears at the end of the paper Received: 23 March 2020 Accepted: 22 April 2020 A newly described coronavirus named severe acute respiratory syndrome Published online: 30 April 2020 coronavirus 2 (SARS-CoV-2), which is the causative agent of coronavirus disease 2019 (COVID-19), has infected over 2.3 million people, led to the death of more than Check for updates 160,000 individuals and caused worldwide social and economic disruption1,2. There are no antiviral drugs with proven clinical efcacy for the treatment of COVID-19, nor are there any vaccines that prevent infection with SARS-CoV-2, and eforts to develop drugs and vaccines are hampered by the limited knowledge of the molecular details of how SARS-CoV-2 infects cells. Here we cloned, tagged and expressed 26 of the 29 SARS-CoV-2 proteins in human cells and identifed the human proteins that physically associated with each of the SARS-CoV-2 proteins using afnity-purifcation mass spectrometry, identifying 332 high-confdence protein–protein interactions between SARS-CoV-2 and human proteins. Among these, we identify 66 druggable human proteins or host factors targeted by 69 compounds (of which, 29 drugs are approved by the US Food and Drug Administration, 12 are in clinical trials and 28 are preclinical compounds). We screened a subset of these in multiple viral assays and found two sets of pharmacological agents that displayed antiviral activity: inhibitors of mRNA translation and predicted regulators of the sigma-1 and sigma-2 receptors. -

Genome–Wide Binding of Transcription Factor ZEB1 in Triple‐Negative
Received: 21 December 2017 | Accepted: 30 March 2018 DOI: 10.1002/jcp.26634 ORIGINAL RESEARCH ARTICLE Genome–wide binding of transcription factor ZEB1 in triple-negative breast cancer cells Varun Maturi1 | Stefan Enroth2 | Carl-Henrik Heldin1 | Aristidis Moustakas1 1 Department of Medical Biochemistry and Microbiology, Science for Life Laboratory, and Zinc finger E-box binding homeobox 1 (ZEB1) is a transcriptional regulator involved in Ludwig Institute for Cancer Research, Uppsala embryonic development and cancer progression. ZEB1 induces epithelial-mesenchy- University, Uppsala, Sweden – 2 Department of Immunology, Genetics and mal transition (EMT). Triple negative human breast cancers express high ZEB1 mRNA Pathology, Science for Life Laboratory, levels and exhibit features of EMT. In the human triple–negative breast cancer cell Uppsala University, Uppsala, Sweden model Hs578T, ZEB1 associates with almost 2,000 genes, representing many cellular Correspondence functions, including cell polarity regulation (DLG2 and FAT3). By introducing a CRISPR- Aristidis Moustakas, Department of Medical Biochemistry and Microbiology, Science for Cas9-mediated 30 bp deletion into the ZEB1 second exon, we observed reduced Life Laboratory, Uppsala University, SE-751 migratory and anchorage-independent growth capacity of these tumor cells. 23 Uppsala, Sweden. Email: [email protected] Transcriptomic analysis of control and ZEB1 knockout cells, revealed 1,372 differentially expressed genes. The TIMP metallopeptidase inhibitor 3 and the teneurin Funding information Vetenskapsrådet, Grant numbers: 2015- transmembrane protein 2 genes showed increased expression upon loss of ZEB1, 02757, K2013-66X-14936-10-5; Ludwig possibly mediating pro-tumorigenic actions of ZEB1. This work provides a resource for Cancer Research, Grant number: Uppsala grant; Cancerfonden, Grant numbers: CAN regulators of cancer progression that function under the transcriptional control of 2012/438, CAN 2015/438 to AM, CAN ZEB1. -
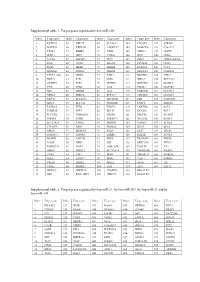
Target Genes Regulated by Hsa-Mir-21, by Hsa-Mir-203, by Hsa-Mir-21 and by Hsa-Mir-143
Supplemental table 1: Target genes regulated by hsa-miR-205 Index Target gene Index Target gene Index Target gene Index Target gene Index Target gene 1 KCTD20 35 UBE2Z 69 SLC38A1 103 LPCAT1 137 STK38L 2 MAPK14 36 YWHAH 70 ANGPTL7 104 MARCKS 138 C1orf123 3 TXNL1 37 RBBP4 71 CTGF 105 MED13 139 GUCD1 4 SPDL1 38 LRP1 72 CYR61 106 IPO7 140 CDK6 5 TCF20 39 IMPAD1 73 TP73 107 PHC2 141 CDKN2AIPNL 6 RAN 40 GNAS 74 EGLN2 108 PICALM 142 CLIP1 7 RGS6 41 MED1 75 ERBB2 109 PLAGL2 143 CUL5 8 HOXA11 42 INPPL1 76 PRRG4 110 NDUFA4 144 C6orf201 9 PAPPA-AS1 43 DDX5 77 F2RL2 111 NDUFB2 145 VTI1A 10 PRR15 44 E2F1 78 GOT1 112 NIPA2 146 SLC5A12 11 ACTRT3 45 E2F5 79 NUFIP2 113 NOTCH2 147 MAML2 12 YES1 46 ZEB2 80 IL24 114 PANK1 148 MAP3K9 13 SRC 47 ERBB3 81 IL32 115 PARD6B 149 NUDT21 14 NPRL3 48 PRKCE 82 RNF217 116 TMEM66 150 DNAJA1 15 NFAT5 49 SLC41A1 83 ZNF585B 117 EZR 151 CCDC108 16 XPOT 50 SLC7A2 84 SIGMAR1 118 ENPP4 152 SHISA6 17 KCTD16 51 ZEB1 85 VEGFA 119 LRRTM4 153 ACP1 18 TMSB4X 52 PHF8 86 BCL9L 120 KCNJ10 154 BCL2 19 PLCXD2 53 TMEM201 87 CREB1 121 PHLPP2 155 NCAPG 20 TNFSF8 54 PTPRJ 88 SERINC3 122 YEATS2 156 KLHL5 21 SLC25A25 55 ETNK1 89 HMGB3 123 VAMP1 157 ACSL4 22 C11orf74 56 XPR1 90 SRD5A1 124 RTN3 158 BCL6 23 GM2A 57 MRPL44 91 PTEN 125 RFX7 159 ITGA5 24 SMNDC1 58 TM9SF2 92 ESRRG 126 RAP2B 160 ACSL1 25 BAMBI 59 PAIP2B 93 PRLR 127 TRAF3IP1 161 EID2B 26 LCOR 60 NEK9 94 ICK 128 SERTAD2 162 TEX35 27 TMEM239 61 NOX5 95 LOH12CR1 129 TOLLIP 163 YY1 28 AMOT 62 DMXL2 96 SLC39A14 130 TMEM55B 164 SMAD1 29 CDK1 63 ETF1 97 BDP1 131 TMEM123 165 SMAD4 30 SQLE 64 -

Molecular Mechanisms Controlling Centriole Duplication
Molecular Mechanisms Controlling Centriole Duplication Item Type text; Electronic Dissertation Authors Boese, Cody Citation Boese, Cody. (2020). Molecular Mechanisms Controlling Centriole Duplication (Doctoral dissertation, University of Arizona, Tucson, USA). Publisher The University of Arizona. Rights Copyright © is held by the author. Digital access to this material is made possible by the University Libraries, University of Arizona. Further transmission, reproduction, presentation (such as public display or performance) of protected items is prohibited except with permission of the author. Download date 05/10/2021 17:34:06 Link to Item http://hdl.handle.net/10150/648667 MOLECULAR MECHANISMS CONTROLLING CENTRIOLE DUPLICATION by Cody Boese ____________________________ Copyright © Cody Boese 2020 A Dissertation Submitted to the Faculty of the GRADUATE INTERDISCIPLINARY PROGRAM IN CANCER BIOLOGY In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY In the Graduate College THE UNIVERSITY OF ARIZONA 2020 THE UNIVERSITY OF ARIZONA GRADUATE COLLEGE As members of the Dissertation Committee, we certify that we have read the dissertation prepared by: Cody Boese titled: Molecular mechanisms controlling centriole assembly and recommend that it be accepted as fulfilling the dissertation requirement for the Degree of Doctor of Philosophy. Gregory C Rogers Date: Jul 21, 2020 Gregory C Rogers Date: Jul 21, 2020 Anne E Cress Date: Oct 22, 2020 Keith Maggert Date: Jul 21, 2020 Ghassan Mouneimne Date: Aug 3, 2020 Nathan Ellis Final approval and acceptance of this dissertation is contingent upon the candidate’s submission of the final copies of the dissertation to the Graduate College. I hereby certify that I have read this dissertation prepared under my direction and recommend that it be accepted as fulfilling the dissertation requirement.