Mechanisms and Models of Cardiac Excitation-Contraction Coupling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Back-To-Basics: the Intricacies of Muscle Contraction
Back-to- MIOTA Basics: The CONFERENCE OCTOBER 11, Intricacies 2019 CHERI RAMIREZ, MS, of Muscle OTRL Contraction OBJECTIVES: 1.Review the anatomical structure of a skeletal muscle. 2.Review and understand the process and relationship between skeletal muscle contraction with the vital components of the nervous system, endocrine system, and skeletal system. 3.Review the basic similarities and differences between skeletal muscle tissue, smooth muscle tissue, and cardiac muscle tissue. 4.Review the names, locations, origins, and insertions of the skeletal muscles found in the human body. 5.Apply the information learned to enhance clinical practice and understanding of the intricacies and complexity of the skeletal muscle system. 6.Apply the information learned to further educate clients on the importance of skeletal muscle movement, posture, and coordination in the process of rehabilitation, healing, and functional return. 1. Epithelial Four Basic Tissue Categories 2. Muscle 3. Nervous 4. Connective A. Loose Connective B. Bone C. Cartilage D. Blood Introduction There are 3 types of muscle tissue in the muscular system: . Skeletal muscle: Attached to bones of skeleton. Voluntary. Striated. Tubular shape. Cardiac muscle: Makes up most of the wall of the heart. Involuntary. Striated with intercalated discs. Branched shape. Smooth muscle: Found in walls of internal organs and walls of vascular system. Involuntary. Non-striated. Spindle shape. 4 Structure of a Skeletal Muscle Skeletal Muscles: Skeletal muscles are composed of: • Skeletal muscle tissue • Nervous tissue • Blood • Connective tissues 5 Connective Tissue Coverings Connective tissue coverings over skeletal muscles: .Fascia .Tendons .Aponeuroses 6 Fascia: Definition: Layers of dense connective tissue that separates muscle from adjacent muscles, by surrounding each muscle belly. -
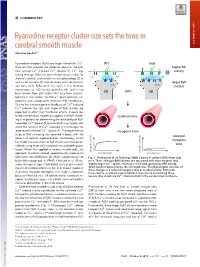
Ryanodine Receptor Cluster Size Sets the Tone in Cerebral Smooth Muscle
COMMENTARY Ryanodine receptor cluster size sets the tone in cerebral smooth muscle COMMENTARY Christian Soellera,1 + Ryanodine receptors (RyRs) are large intracellular Ca2 WT mdx channels that provide the molecular basis of the pro- K+ K+ higher BK + + + cess termed Ca2 -induced Ca2 release (1). Ca2 sig- activity naling through RyRs has been shown to be critical for CaC BK BK skeletal, cardiac, and smooth muscle physiology (2) as well as for neurons (3) and secretory cells like pancre- larger RyR atic beta cells. RyRs were first seen in the electron RyRs RyRs clusters microscope as ∼30-nm-size particles (4), and it had SMCs been known from EM studies that they form clusters, SR SR typically in the narrow “junctional” space between sar- colemma and sarcoplasmic reticulum (SR) membranes. + Due to the inherent positive feedback of Ca2 -induced + Ca2 release, the size and shape of RyR clusters are expected to affect their functional activity. Indeed, de- SMCs SMCs tailed mathematical modeling suggests that RyR cluster- cerebral artery ing is important for determining the excitability of RyR- + mediated Ca2 release (5, 6) and could, in principle, also + affect the amount of Ca2 released in microscopic re- 2+ lease events termed Ca sparks (7). The experimental myogenic tone study of RyR clustering has received a boost with the reduced advent of optical superresolution microscopy, which myogenic has made the assessment of RyR cluster size more acces- sible by using essentially standard immunolabeling pro- tone tocols. When first applied in cardiac muscle cells, this myogenic tone (%) tone myogenic approach revealed a broad, approximately exponential vessel pressure (%) tone myogenic vessel pressure RyR cluster size distribution (8), which had previously not Fig. -

Structure and Function of the Human Ryanodine Receptors and Their Association with Myopathies—Present State, Challenges, and Perspectives
molecules Review Structure and Function of the Human Ryanodine Receptors and Their Association with Myopathies—Present State, Challenges, and Perspectives Vladena Bauerová-Hlinková * , Dominika Hajdúchová † and Jacob A. Bauer Institute of Molecular Biology, Slovak Academy of Sciences, Dúbravská Cesta 21, 845 51 Bratislava, Slovakia; [email protected] (D.H.); [email protected] (J.A.B.) * Correspondence: [email protected]; Tel.: +421-2-5930-7439 † Current address: Department of Pathological Physiology, Jessenius Faculty of Medicine in Martin, Comenius University in Bratislava, Malá Hora 4C, 036 01 Martin, Slovakia. Academic Editor: Jacopo Sgrignani and Giovanni Grazioso Received: 31 July 2020; Accepted: 30 August 2020; Published: 4 September 2020 Abstract: Cardiac arrhythmias are serious, life-threatening diseases associated with the dysregulation of Ca2+ influx into the cytoplasm of cardiomyocytes. This dysregulation often arises from dysfunction of ryanodine receptor 2 (RyR2), the principal Ca2+ release channel. Dysfunction of RyR1, the skeletal muscle isoform, also results in less severe, but also potentially life-threatening syndromes. The RYR2 and RYR1 genes have been found to harbor three main mutation “hot spots”, where mutations change the channel structure, its interdomain interface properties, its interactions with its binding partners, or its dynamics. In all cases, the result is a defective release of Ca2+ ions from the sarcoplasmic reticulum into the myocyte cytoplasm. Here, we provide an overview of the most frequent diseases resulting from mutations to RyR1 and RyR2, briefly review some of the recent experimental structural work on these two molecules, detail some of the computational work describing their dynamics, and summarize the known changes to the structure and function of these receptors with particular emphasis on their N-terminal, central, and channel domains. -

Muscular System ANS 215 Physiology and Anatomy of Domesticated Animals
Muscular System ANS 215 Physiology and Anatomy of Domesticated Animals I. Skeletal Muscle Contraction A. Neuromuscular junction functions as an amplifier for a nerve impulse B. Arrival of a spinal or cranial nerve impulse at the neuromuscular junction results in release of acetylcholine (Ach) into the space between the nerve fiber terminal branch and the muscle fiber C. Release of Ach is accelerated because Ca ions from extracellular fluid enter the prejunctional membrane when the nerve impulse arrives D. Ach is the stimulus that increases the permeability of the muscle fiber membrane for Na ions, after which depolarization begins E. Depolarization proceeds in all directions from the neuromuscular junction F. Impulse is conducted into all parts of the muscle fiber by the sarcotubular system (synchronizes muscle fiber contraction) G. Low concentration of Ca in the extracellular fluid is recognized clinically in dairy cows after calving (parturient paresis) as a state of semi-paralysis caused by partial neuromuscular block. H. Almost immediately after its release Ach is hydrolyzed by the enzyme acetylcholinesterase into acetic acid and choline I. Next depolarization must await the arrival of the next nerve impulse J. Tubules of sarcoplasmic reticulum have a relatively high concentration of Ca ions K. Depolarization of these tubules results in a simultaneous release of Ca ions into the sarcoplasm, which in turn diffuse rapidly into the myofibrils L. Presence of Ca ions within the myofibrils initiates the contraction process M. The Ca ions are returned rapidly by active transport to the sarcoplasmic reticulum after contraction is initiated and are released again when the next signal arrives. -

Histology of Muscle Tissue
HISTOLOGY OF MUSCLE TISSUE Dr. Sangeeta Kotrannavar Assistant Professor Dept. of Anatomy, USM-KLE IMP, Belagavi Objectives Distinguish the microscopic features of • Skeletal • Cardiac • Smooth muscles Muscle • Latin musculus =little mouse (mus) • Muscle cells are known as MYOCYTES. • Myocytes are elongated so referred as muscle fibers Fleshy • Definition • Muscle is a contractile tissue which brings about movements Tendons Muscle makes up 30-35% (in women) & 40-45% (in men) of body mass Type of muscles BASED ON BASED ON BASED ON LOCATION STRIATIONS CONTROL Skeletal / Somatic STRIATED / STRIPED VOLUNTARY Smooth / Visceral UN-STRIATED / IN-VOLUNTARY UNSTRIPED Cardiac STRIATED / STRIPED IN-VOLUNTARY SKELETAL MUSCLE Skeletal muscle organization Muscles are complex structures: arranged in fascicles Muscle bundles / fascicles • Epimysium surrounds entire muscle – Dense CT that merges with tendon – Epi = outer, Mys = muscle • Perimysium surrounds muscle fascicles – Peri = around – Within a muscle fascicle are many muscle fibers • Endomysium surrounds muscle fiber – Endo = within SKELETAL MUSCLE • Each bundles contains many muscle fiber Structure of a skeletal muscle fiber • Elongated, unbranched cylindrical fibers • Length- 1 mm – 5 cm, Width – 10 mm - 100μm • Fibers have striations of dark & light bands • Many flat nuclei beneath sarcolemma • Plasma membrane = sarcolemma • Smooth endoplsmic reticulum = sarcoplasmic reticulum (SR) • Cytoplasm = sarcoplasm • Mitochondria = sarcosomes • Each muscle fiber made of long cylindrical myofibrils Structure -

Muscle Contraction
Muscle Physiology Dr. Ebneshahidi © 2009 Ebneshahidi Skeletal Muscle Figure 9.2 (a) © 2009 Ebneshahidi Functions of the muscular system . 1. Locomotion – body movements are due to skeletal muscle contraction. 2. Vasoconstriction and vasodilatation - constriction and dilation of blood vessel walls are the results of smooth muscle contraction. 3. Peristalsis – wavelike motion along the digestive tract is produced by the smooth muscle. 4. Cardiac motion – heart chambers pump blood to the lungs and to the body because of cardiac muscle contraction. 5. Posture maintenance - contraction of skeletal muscles maintains body posture and muscle tone. 6. Heat generation – about 75% of ATP energy used in muscle contraction is released as heat. © 2009 Ebneshahidi Comparison of the three types of muscle . Striation: only present in skeletal and cardiac muscles. Absent in smooth muscle. Nucleus: smooth and cardiac muscles are uninculcated (one nucleus per cell) skeletal muscle is multinucleated (several nuclei per cell). Transverse tubule (T tubule): well developed in skeletal and cardiac muscles to transport calcium. Absent in smooth muscle. Intercalated disk: specialized intercellular junction that only occurs in cardiac muscle. Control: skeletal muscle is always under voluntary control‚ with some exceptions (the tongue and pili arrector muscles in the dermis). Smooth and cardiac muscles are under involuntary control. © 2009 Ebneshahidi Innervation: motor unit . a) a motor nerve and a myofibril from a neuromuscular junction where gap (called synapse) occurs between the two structures. At the end of motor nerve‚ neurotransmitter (i.e. acetylcholine) is stored in synaptic vesicles which will release the neurotransmitter using exocytosis upon the stimulation of a nerve impulse. -

THE SARCOPLASMIC RETICULUM, the T SYSTEM, and the MOTOR TERMINALS of SLOW and TWITCH MUSCLE FIBERS in the GARTER SNAKE Defined F
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by PubMed Central THE SARCOPLASMIC RETICULUM, THE T SYSTEM, AND THE MOTOR TERMINALS OF SLOW AND TWITCH MUSCLE FIBERS IN THE GARTER SNAKE ARTHUR HESS From the Department of Physiology, University of Utah College of Medicine, Salt Lake City, Utah ABSTRACT Twitch and slow muscle fibers, identified morphologically in the garter snake, have been examined in the electron microscope. The transverse tubular system and the sarcoplasmic reticulum are separate entities distinct from each other. In twitch fibers, the tubular system and the dilated sacs of the sarcoplasmic reticulum form triads at the level of junction of A and I bands. In the slow fibers, the sarcoplasmic reticulum is severely depleted in amount and the transverse tubular system is completely absent. The junctional folds of the post- synaptic membrane of the muscle fiber under an "en grappe" ending of a slow fiber arc not so frequent or regular in occurrence or so wide or so long as under the "en plaque" ending of a twitch fiber. Some physiological implications of these differences in fine struc- ture of twitch and slow fibers are discussed. The absence of the transverse tubular system and reduction in amount of sarcoplasmic reticulum, along with the consequent disposition of the fibrils, the occurrence of multiple nerve terminals, and the degree of complexity of the post junctional folds of the sarcolemma appear to be the morphological basis for the physiological reaction of slow muscle fibers. Extrafusal muscle fibers may be classified both irregular distribution of fibrils and multiple motor morphologically and physiologically as twitch endings (5). -

Levels of Muscle Structure
Levels of Muscle Structure • Muscle • Muscle Bundle • Muscle Fiber (myofiber) • Myofibrils • Myofilaments 1 Muscle Muscle Bundle • Contains 10-20 myofibers • Encased by perimysium • Can see with the naked eye Myofiber (Muscle fiber) • Individual muscle cell • Multinucleated • Encased by endomysium • Cell wall: sarcolemma 2 Myofibrils • Embedded in sarcoplasm • Mitochondria located between myofibrils Myofibrils • Comprised of repeating units: sarcomeres – A band – I band –Z disk –H zone – Pseudo-H zone 3 Sarcomere Sarcomere Myofilaments • Contractile Proteins – Myosin – Actin • Regulatory Proteins – Tropomyosin – Troponin • Structural Proteins – Z – Line Proteins 4 Contractile Proteins • Myosin – 70 – 80% of the total protein – Thick filament – Burns the ATP for muscle contraction Myosin & Its Fragments Chymotrypsin – Myosin head moves S1 back and forth to perform a muscle ROD Alkali Reg ula to r y li g ht c h ain - light chain COO contraction LMM COO- Actin binding and A TPa se activity HMM Trypsin Contractile Proteins • Actin – 20% of the myofibrillar protein – Thin Filament – Globular protein (G- protein) – Arranged like a twisted pearl necklace (F-protein) – Myosin head attaches to the Actin Regulatory Proteins • Regulate contraction and the speed of contraction • Tropomyosin • Troponin 5 Regulatory Proteins • Tropomyosin – Thin protein that lays around the Actin proteins AA End View Top View Regulatory Proteins • Troponin • 3 Subunits – TnT • Binds tropomyosin – TnI • Inhibitory subunit – TnC • Ca2+ binding subunit 3 Dimensional 6 -

Modulation of Ryanodine Receptor Ca2+ Channels (Review)
MOLECULAR MEDICINE REPORTS 3: 199-204, 2010 199 Modulation of ryanodine receptor Ca2+ channels (Review) TERUTAKA OZAWA Department of Physiology, Tohoku University Graduate School of Medicine, Sendai 980-8575, Japan Received October 9, 2009; Accepted December 28, 2009 DOI: 10.3892/mmr_00000240 Abstract. Ryanodine-sensitive Ca2+ release channels deals with the modulation mechanisms of RyR proteins by (ryanodine receptors, RyRs) play a crucial role in the associated proteins and phosphorylation. mobilization of Ca2+ from the sarcoplasmic reticulum (SR) during the excitation-contraction coupling of muscle cells. In skeletal muscle, depolarization of transverse tubules Contents activates the RyR, whereas in cardiac muscle, a Ca2+ influx through an L-type Ca2+ channel activates the RyR. The RyR 1. Introduction is also activated by caffeine, a low concentration (<10 µM) 2. Activation of the ryanodine receptor of ryanodine or cyclic ADP-ribose. RyR activity is inhibited 3. Modulation of the ryanodine receptor by Mg2+, ruthenium red, or higher concentrations (≥100 µM) 4. Conclusion of ryanodine. The activity of RyR channels is modulated by phosphorylation and by associated proteins, including calmodulin (CaM), calsequestrin (CSQ) and FK506-binding 1. Introduction proteins (FKBPs). In muscle cells, apoCaM (Ca2+-free CaM) activates the RyR channel, and Ca2+ CaM (Ca2+-bound CaM) In many cell types, intracellular Ca2+ stores play an essential 2+ 2+ inhibits the channel. CSQ can bind approximately 40 moles role in the regulation of cytosolic Ca concentration ([Ca ]i), of Ca2+/mole of CSQ in the SR lumen of muscle cells, and the elevation of which triggers various cellular events, including interacts functionally with RyR protein. -

Muscles and Muscle Tissue 279
278 UNIT 2 Covering, Support, and Movement of the Body Muscles and WHY THIS 9 Muscle Tissue MATTERS In this chapter, you will learn that Muscles use actin and myosin molecules to convert the energy of ATP into force beginning with 9.1 Overview of muscle types, special characteristics, next exploring and functions then exploring Skeletal muscle Smooth muscle and investigating then asking and asking 9.2 Gross and 9.4 How does a nerve 9.9 How does smooth muscle microscopic anatomy impulse cause a muscle fiber differ from skeletal muscle? to contract? and and finally, exploring and 9.3 Intracellular structures and sliding 9.5 What are the properties of Developmental Aspects filament model whole muscle contraction? of Muscles and 9.6 How do muscles generate ATP? and 9.7 What determines the force, velocity, and duration of contraction? and 9.8 How does skeletal muscle respond to exercise? Electron micrograph of a bundle of skeletal muscle fibers wrapped in connective tissue. < 278 Chapter 9 Muscles and Muscle Tissue 279 ecause flexing muscles look like mice scurrying beneath the by the nervous system. Most of us have no conscious control B skin, some scientist long ago dubbed them muscles, from over how fast our heart beats. the Latin mus meaning “little mouse.” Indeed, we tend to ● Key words to remember for cardiac muscle are cardiac, stri- think of the rippling muscles of professional boxers or weight lift- ated, and involuntary. ers when we hear the word muscle. But muscle is also the domi- nant tissue in the heart and in the walls of other hollow organs. -

Fine Structure and Organization of the Sarcoplasmic Reticulum in the Sino-Auricular Fibres of Frog Heart Michel Duvert and Andre L
Fine Structure and Organization of the Sarcoplasmic Reticulum in the Sino-Auricular Fibres of Frog Heart Michel Duvert and Andre L. Barets Laboratoire de Cytologie, Universite de Bordeaux 2, Avenue des Facultes, F-33405 Talence- Cedex-France Z. Naturforsch. 34 c, 865 - 875 (1979); received May 25, 1979 Cardiac Muscle, Excitation-Contraction Coupling, Sarcoplasmic Reticulum, In Situ SR Mor phology, SR-Mitochondria Relationship The ultrastructure of the sarcoplasmic reticulum (SR) of the sino-auricular fibres of frog heart has been investigated especially in order to evaluate its possible relationship with several cellular organelles. The SR appears as a complex unit which can be divided into three regions: — peripheral SR, localized beneath the sarcolemma. It is composed of junctional SR, which forms specialized complexes (couplings) with the sarcolemma, and peripheral SR s. str. which fre quently extends its anastomotic tubules around the caveolae. — juxtafibrillar SR, in relation with myofibrils, at the level of Z lines and I bands. — intermediary SR which is the most abundant. It contains dense granules, able to bind stronti um (and calcium) and, from place to place, is formed into flattened saccules bearing fenestrations where microfilaments frequently pass through. It allows peripheral SR and juxtafibrillar SR to communicate, giving unity to the reticular system and, on the other hand, shows close topographi cal relations with mitochondria. The great extension of the SR, its heterogeneity and its relationships with several cellular -
New Insights Into Cardiac Excitation- Contraction Coupling in Normal and Hypertension/Failure Animal Models
Journal of Human Hypertension (1997) 11, 555–558 1997 Stockton Press. All rights reserved 0950-9240/97 $12.00 New insights into cardiac excitation- contraction coupling in normal and hypertension/failure animal models MB Cannell Department of Pharmacology & Clinical Pharmacology, St. George’s Hospital Medical School, Cranmer Terrace, London SW17 ORE, UK Application of the confocal microscope to enzymatically mal (or well ‘tuned’) for activating calcium sparks which isolated cardiac myocytes has revealed that excitation- will minimise the requirement for calcium influx into the contraction coupling is a ‘local control phenomenon’. cell. In the spontaneous hypertensive rat (SHR) model of The whole cell calcium transient is made up of the tem- hypertension, the relationship between calcium channel poral and spatial summation of a large number of micro- activity and calcium release from internal stores is alt- scopic calcium release events called ‘calcium sparks’. ered in a way that results in a reduced contraction The opening of a single calcium channel in the surface strength. The relationship between the calcium channel b membrane can activate a calcium spark and there is a current and Ps is restored by -adrenergic stimulation non-linear relationship between the amplitude of the sin- in the hypertrophy model but not in hearts which are gle calcium channel flux and the probability of activat- failing. These results suggest that a novel approach to ing a calcium spark (Ps). Mathematical modelling shows treating certain types of heart failure could be to modify that the relationship between surface membrane cal- the gating behaviour of the sarcolemmal calcium chan- cium channel gating and the activation of calcium nel to ‘retune’ ability of the sarcolemmal calcium chan- release channels in internal stores is very sensitive to nels to activate calcium release from internal stores, the geometric relationship between these channels.