Crystal Structures of Wild Type and Disease Mutant Forms of the Ryanodine Receptor SPRY2 Domain
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Mineralocorticoid Receptor Leads to Increased Expression of EGFR
www.nature.com/scientificreports OPEN The mineralocorticoid receptor leads to increased expression of EGFR and T‑type calcium channels that support HL‑1 cell hypertrophy Katharina Stroedecke1,2, Sandra Meinel1,2, Fritz Markwardt1, Udo Kloeckner1, Nicole Straetz1, Katja Quarch1, Barbara Schreier1, Michael Kopf1, Michael Gekle1 & Claudia Grossmann1* The EGF receptor (EGFR) has been extensively studied in tumor biology and recently a role in cardiovascular pathophysiology was suggested. The mineralocorticoid receptor (MR) is an important efector of the renin–angiotensin–aldosterone‑system and elicits pathophysiological efects in the cardiovascular system; however, the underlying molecular mechanisms are unclear. Our aim was to investigate the importance of EGFR for MR‑mediated cardiovascular pathophysiology because MR is known to induce EGFR expression. We identifed a SNP within the EGFR promoter that modulates MR‑induced EGFR expression. In RNA‑sequencing and qPCR experiments in heart tissue of EGFR KO and WT mice, changes in EGFR abundance led to diferential expression of cardiac ion channels, especially of the T‑type calcium channel CACNA1H. Accordingly, CACNA1H expression was increased in WT mice after in vivo MR activation by aldosterone but not in respective EGFR KO mice. Aldosterone‑ and EGF‑responsiveness of CACNA1H expression was confrmed in HL‑1 cells by Western blot and by measuring peak current density of T‑type calcium channels. Aldosterone‑induced CACNA1H protein expression could be abrogated by the EGFR inhibitor AG1478. Furthermore, inhibition of T‑type calcium channels with mibefradil or ML218 reduced diameter, volume and BNP levels in HL‑1 cells. In conclusion the MR regulates EGFR and CACNA1H expression, which has an efect on HL‑1 cell diameter, and the extent of this regulation seems to depend on the SNP‑216 (G/T) genotype. -

List of Genes Associated with Sudden Cardiac Death (Scdgseta) Gene
List of genes associated with sudden cardiac death (SCDgseta) mRNA expression in normal human heart Entrez_I Gene symbol Gene name Uniprot ID Uniprot name fromb D GTEx BioGPS SAGE c d e ATP-binding cassette subfamily B ABCB1 P08183 MDR1_HUMAN 5243 √ √ member 1 ATP-binding cassette subfamily C ABCC9 O60706 ABCC9_HUMAN 10060 √ √ member 9 ACE Angiotensin I–converting enzyme P12821 ACE_HUMAN 1636 √ √ ACE2 Angiotensin I–converting enzyme 2 Q9BYF1 ACE2_HUMAN 59272 √ √ Acetylcholinesterase (Cartwright ACHE P22303 ACES_HUMAN 43 √ √ blood group) ACTC1 Actin, alpha, cardiac muscle 1 P68032 ACTC_HUMAN 70 √ √ ACTN2 Actinin alpha 2 P35609 ACTN2_HUMAN 88 √ √ √ ACTN4 Actinin alpha 4 O43707 ACTN4_HUMAN 81 √ √ √ ADRA2B Adrenoceptor alpha 2B P18089 ADA2B_HUMAN 151 √ √ AGT Angiotensinogen P01019 ANGT_HUMAN 183 √ √ √ AGTR1 Angiotensin II receptor type 1 P30556 AGTR1_HUMAN 185 √ √ AGTR2 Angiotensin II receptor type 2 P50052 AGTR2_HUMAN 186 √ √ AKAP9 A-kinase anchoring protein 9 Q99996 AKAP9_HUMAN 10142 √ √ √ ANK2/ANKB/ANKYRI Ankyrin 2 Q01484 ANK2_HUMAN 287 √ √ √ N B ANKRD1 Ankyrin repeat domain 1 Q15327 ANKR1_HUMAN 27063 √ √ √ ANKRD9 Ankyrin repeat domain 9 Q96BM1 ANKR9_HUMAN 122416 √ √ ARHGAP24 Rho GTPase–activating protein 24 Q8N264 RHG24_HUMAN 83478 √ √ ATPase Na+/K+–transporting ATP1B1 P05026 AT1B1_HUMAN 481 √ √ √ subunit beta 1 ATPase sarcoplasmic/endoplasmic ATP2A2 P16615 AT2A2_HUMAN 488 √ √ √ reticulum Ca2+ transporting 2 AZIN1 Antizyme inhibitor 1 O14977 AZIN1_HUMAN 51582 √ √ √ UDP-GlcNAc: betaGal B3GNT7 beta-1,3-N-acetylglucosaminyltransfe Q8NFL0 -

Transcriptomic Analysis of Native Versus Cultured Human and Mouse Dorsal Root Ganglia Focused on Pharmacological Targets Short
bioRxiv preprint doi: https://doi.org/10.1101/766865; this version posted September 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Transcriptomic analysis of native versus cultured human and mouse dorsal root ganglia focused on pharmacological targets Short title: Comparative transcriptomics of acutely dissected versus cultured DRGs Andi Wangzhou1, Lisa A. McIlvried2, Candler Paige1, Paulino Barragan-Iglesias1, Carolyn A. Guzman1, Gregory Dussor1, Pradipta R. Ray1,#, Robert W. Gereau IV2, # and Theodore J. Price1, # 1The University of Texas at Dallas, School of Behavioral and Brain Sciences and Center for Advanced Pain Studies, 800 W Campbell Rd. Richardson, TX, 75080, USA 2Washington University Pain Center and Department of Anesthesiology, Washington University School of Medicine # corresponding authors [email protected], [email protected] and [email protected] Funding: NIH grants T32DA007261 (LM); NS065926 and NS102161 (TJP); NS106953 and NS042595 (RWG). The authors declare no conflicts of interest Author Contributions Conceived of the Project: PRR, RWG IV and TJP Performed Experiments: AW, LAM, CP, PB-I Supervised Experiments: GD, RWG IV, TJP Analyzed Data: AW, LAM, CP, CAG, PRR Supervised Bioinformatics Analysis: PRR Drew Figures: AW, PRR Wrote and Edited Manuscript: AW, LAM, CP, GD, PRR, RWG IV, TJP All authors approved the final version of the manuscript. 1 bioRxiv preprint doi: https://doi.org/10.1101/766865; this version posted September 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Back-To-Basics: the Intricacies of Muscle Contraction
Back-to- MIOTA Basics: The CONFERENCE OCTOBER 11, Intricacies 2019 CHERI RAMIREZ, MS, of Muscle OTRL Contraction OBJECTIVES: 1.Review the anatomical structure of a skeletal muscle. 2.Review and understand the process and relationship between skeletal muscle contraction with the vital components of the nervous system, endocrine system, and skeletal system. 3.Review the basic similarities and differences between skeletal muscle tissue, smooth muscle tissue, and cardiac muscle tissue. 4.Review the names, locations, origins, and insertions of the skeletal muscles found in the human body. 5.Apply the information learned to enhance clinical practice and understanding of the intricacies and complexity of the skeletal muscle system. 6.Apply the information learned to further educate clients on the importance of skeletal muscle movement, posture, and coordination in the process of rehabilitation, healing, and functional return. 1. Epithelial Four Basic Tissue Categories 2. Muscle 3. Nervous 4. Connective A. Loose Connective B. Bone C. Cartilage D. Blood Introduction There are 3 types of muscle tissue in the muscular system: . Skeletal muscle: Attached to bones of skeleton. Voluntary. Striated. Tubular shape. Cardiac muscle: Makes up most of the wall of the heart. Involuntary. Striated with intercalated discs. Branched shape. Smooth muscle: Found in walls of internal organs and walls of vascular system. Involuntary. Non-striated. Spindle shape. 4 Structure of a Skeletal Muscle Skeletal Muscles: Skeletal muscles are composed of: • Skeletal muscle tissue • Nervous tissue • Blood • Connective tissues 5 Connective Tissue Coverings Connective tissue coverings over skeletal muscles: .Fascia .Tendons .Aponeuroses 6 Fascia: Definition: Layers of dense connective tissue that separates muscle from adjacent muscles, by surrounding each muscle belly. -

Stac Adaptor Proteins Regulate Trafficking and Function of Muscle
Stac adaptor proteins regulate trafficking and function + of muscle and neuronal L-type Ca2 channels Alexander Polster, Stefano Perni, Hicham Bichraoui, and Kurt G. Beam1 Department of Physiology and Biophysics, University of Colorado Anschutz Medical Campus, Aurora, CO 80045 Contributed by Kurt G. Beam, December 5, 2014 (sent for review November 11, 2014; reviewed by Bruce P. Bean and Terry P. Snutch) + Excitation–contraction (EC) coupling in skeletal muscle depends precisely regulated. As for other high-voltage activated Ca2 upon trafficking of CaV1.1, the principal subunit of the dihydropyr- channels, the auxiliary β-subunit is important for membrane traf- + idine receptor (DHPR) (L-type Ca2 channel), to plasma membrane ficking (5, 6). However, it is clear that an additional factor(s) is regions at which the DHPRs interact with type 1 ryanodine recep- also crucial. Specifically, CaV1.1 expresses poorly (7) or not at all tors (RyR1) in the sarcoplasmic reticulum. A distinctive feature of in mammalian cells that are not of muscle origin (e.g., tsA201 2+ this trafficking is that CaV1.1 expresses poorly or not at all in cells), whereas the closely related cardiac/neuronal L-type Ca mammalian cells that are not of muscle origin (e.g., tsA201 cells), channel, CaV1.2, is readily expressed in such cells (8–10). The in which all of the other nine CaV isoforms have been successfully absence of RyR1 seems insufficient to account for the difficulty expressed. Here, we tested whether plasma membrane trafficking of expressing Ca 1.1 in tsA201 cells because Ca 1.1 expresses of Ca 1.1 in tsA201 cells is promoted by the adapter protein Stac3, V V V well in dyspedic myotubes, albeit producing less current per because recent work has shown that genetic deletion of Stac3 in channel (3). -

Identification of Gene Expression and DNA Methylation of SERPINA5 and TIMP1 As Novel Prognostic Markers in Lower-Grade Gliomas
Identification of gene expression and DNA methylation of SERPINA5 and TIMP1 as novel prognostic markers in lower-grade gliomas Wen-Jing Zeng1,2,3,4, Yong-Long Yang5, Zhi-Peng Wen1,2, Peng Chen1,2, Xiao-Ping Chen1,2 and Zhi-Cheng Gong3,4 1 Department of Clinical Pharmacology, Xiangya Hospital, Central South University, Changsha, Hunan, China 2 Institute of Clinical Pharmacology, Central South University, Hunan Key Laboratory of Pharmacogenetics, Changsha, Hunan, China 3 Department of Pharmacy, Xiangya Hospital, Central South University, Changsha, Hunan, China 4 National Clinical Research Center for Geriatric Disorders (XIANGYA), Xiangya Hospital, Central South University, Changsha, Hunan, China 5 Department of Clinical Pharmacology Research Center, Changsha Carnation Geriatrics Hospital, Changsha, Hunan, China ABSTRACT Background. Lower-grade gliomas (LGGs) is characteristic with great difference in prognosis. Due to limited prognostic biomarkers, it is urgent to identify more molecular markers to provide a more objective and accurate tumor classification system for LGGs. Methods. In the current study, we performed an integrated analysis of gene expression data and genome-wide methylation data to determine novel prognostic genes and methylation sites in LGGs. Results. To determine genes that differentially expressed between 44 short-term survivors (<2 years) and 48 long-term survivors (≥2 years), we searched LGGs TCGA RNA-seq dataset and identified 106 differentially expressed genes. SERPINA5 and Submitted 11 October 2019 Accepted 9 May 2020 TIMP1 were selected for further study. Kaplan–Meier plots showed that SERPINA5 Published 3 June 2020 and TIMP1 expression were significantly correlated with overall survival (OS) and Corresponding authors relapse-free survival (RFS) in TCGA LGGs patients. -

RYR3 Gene Polymorphisms and Cardiovascular Disease Outcomes in the Context of Antihypertensive Treatment
The Pharmacogenomics Journal (2013) 13, 330 -- 334 & 2013 Macmillan Publishers Limited All rights reserved 1470-269X/13 www.nature.com/tpj ORIGINAL ARTICLE RYR3 gene polymorphisms and cardiovascular disease outcomes in the context of antihypertensive treatment AI Lynch1, MR Irvin1, E Boerwinkle2, BR Davis3, LK Vaughan4, CE Ford3, B Aissani1, JH Eckfeldt5, DK Arnett1 and S Shrestha1 Nearly one-third of adults in the United States have hypertension, which is associated with increased cardiovascular disease (CVD) morbidity and mortality. The goal of antihypertensive pharmacogenetic research is to enhance understanding of drug response based on the interaction of individual genetic architecture and antihypertensive therapy to improve blood pressure control and ultimately prevent CVD outcomes. In the context of the Genetics of Hypertension Associated Treatment study and using a case-only design, we examined whether single-nucleotide polymorphisms in RYR3 interact with four classes of antihypertensive drugs, particularly the calcium channel blocker amlodipine versus other classes, to modify the risk of coronary heart disease (CHD; fatal CHD and non-fatal myocardial infarction combined) and heart failure (HF) in high-risk hypertensive individuals. RYR3 mediates the mobilization of stored Ca þ 2 in cardiac and skeletal muscle to initiate muscle contraction. There was suggestive evidence of pharmacogenetic effects on HF, the strongest of which was for rs877087, with the smallest P-value ¼ 0.0005 for the codominant model when comparing amlodipine versus all other treatments. There were no pharmacogenetic effects observed for CHD. The findings reported here for the case-only analysis of the antihypertensive pharmacogenetic effect of RYR3 among 3058 CHD cases and 1940 HF cases show that a hypertensive patient’s genetic profile may help predict which medication(s) might better lower CVD risk. -

Ion Channels 3 1
r r r Cell Signalling Biology Michael J. Berridge Module 3 Ion Channels 3 1 Module 3 Ion Channels Synopsis Ion channels have two main signalling functions: either they can generate second messengers or they can function as effectors by responding to such messengers. Their role in signal generation is mainly centred on the Ca2 + signalling pathway, which has a large number of Ca2+ entry channels and internal Ca2+ release channels, both of which contribute to the generation of Ca2 + signals. Ion channels are also important effectors in that they mediate the action of different intracellular signalling pathways. There are a large number of K+ channels and many of these function in different + aspects of cell signalling. The voltage-dependent K (KV) channels regulate membrane potential and + excitability. The inward rectifier K (Kir) channel family has a number of important groups of channels + + such as the G protein-gated inward rectifier K (GIRK) channels and the ATP-sensitive K (KATP) + + channels. The two-pore domain K (K2P) channels are responsible for the large background K current. Some of the actions of Ca2 + are carried out by Ca2+-sensitive K+ channels and Ca2+-sensitive Cl − channels. The latter are members of a large group of chloride channels and transporters with multiple functions. There is a large family of ATP-binding cassette (ABC) transporters some of which have a signalling role in that they extrude signalling components from the cell. One of the ABC transporters is the cystic − − fibrosis transmembrane conductance regulator (CFTR) that conducts anions (Cl and HCO3 )and contributes to the osmotic gradient for the parallel flow of water in various transporting epithelia. -

Table S1. Detailed Clinical Features of Individuals with IDDCA and LADCI Syndromes
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Med Genet Table S1. Detailed clinical features of individuals with IDDCA and LADCI syndromes Lodder E., De Nittis P., Koopman C. et al., 2016 Family A Family B Family C Family D Family E Family F Individual 1 2 3 4 5 6 7 8 9 Gender, Age (years) F, 22 F, 20 F, 6 F, 11 M, 9 F, 12 F, 13 M, 8 M, 23 c.249G>A,r.249_250 c.249G>A,r.249_250 Nucleotide change c.249+1G>T/ c.249+3G>T/ c.249+3G>T/ c.906C>G/ c.242C>T/ c.242C>T/ c.242C>T/ ins249+1_249+25/ ins249+1_249+25/ (NM_006578.3) c.249+1G>T c.249+3G>T c.249+3G>T c.906C>G c.242C>T c.242C>T c.242C>T c.994C>T c.994C>T Amino acid change p.Asp84Valfs*52/ p.Asp84Valfs*52/ p.Asp84Leufs*31/ p.Asp84Valfs*31/ p.Asp84Valfs*31/ p.Tyr302*/ p.(Ser81Leu)/ p.(Ser81Leu)/ p.(Ser81Leu)/ (NP_006569.1) p.(Arg332*) p.(Arg332*) p.Asp84Leufs*31 p.Asp84Valfs*31 p.Asp84Valfs*31 p.Tyr302* p.(Ser81Leu) p.(Ser81Leu) p.(Ser81Leu) 3580 g (50th 2751 g (15 th Birth weight NA NA NA 2845 g (15th NA NA NA percentile) percentile) percentile) Ethnicity Italy Italy Jordan Puerto Rico Puerto Rico India Morocco Morocco Brazil Consanguinity − − + + + − − − + Altered speech + + NR + + + + + NA development - Verbal NA NA nonverbal unremarkable unremarkable NA NA NA NA understanding - Lexical production NA NA nonverbal delayed delayed nonverbal delayed delayed NA Intellectual + + + + + + mild mild mild disability (ID) Epilepsy + + + - -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -

Preclinical Model Systems of Ryanodine Receptor 1-Related Myopathies and Malignant Hyperthermia
Lawal et al. Orphanet Journal of Rare Diseases (2020) 15:113 https://doi.org/10.1186/s13023-020-01384-x REVIEW Open Access Preclinical model systems of ryanodine receptor 1-related myopathies and malignant hyperthermia: a comprehensive scoping review of works published 1990– 2019 Tokunbor A. Lawal1, Emily S. Wires2, Nancy L. Terry3, James J. Dowling4 and Joshua J. Todd1* Abstract Background: Pathogenic variations in the gene encoding the skeletal muscle ryanodine receptor (RyR1) are associated with malignant hyperthermia (MH) susceptibility, a life-threatening hypermetabolic condition and RYR1- related myopathies (RYR1-RM), a spectrum of rare neuromuscular disorders. In RYR1-RM, intracellular calcium dysregulation, post-translational modifications, and decreased protein expression lead to a heterogenous clinical presentation including proximal muscle weakness, contractures, scoliosis, respiratory insufficiency, and ophthalmoplegia. Preclinical model systems of RYR1-RM and MH have been developed to better understand underlying pathomechanisms and test potential therapeutics. Methods: We conducted a comprehensive scoping review of scientific literature pertaining to RYR1-RM and MH preclinical model systems in accordance with the PRISMA Scoping Reviews Checklist and the framework proposed by Arksey and O’Malley. Two major electronic databases (PubMed and EMBASE) were searched without language restriction for articles and abstracts published between January 1, 1990 and July 3, 2019. Results: Our search yielded 5049 publications from which 262 were included in this review. A majority of variants tested in RYR1 preclinical models were localized to established MH/central core disease (MH/CCD) hot spots. A total of 250 unique RYR1 variations were reported in human/rodent/porcine models with 95% being missense substitutions. -
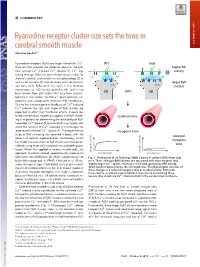
Ryanodine Receptor Cluster Size Sets the Tone in Cerebral Smooth Muscle
COMMENTARY Ryanodine receptor cluster size sets the tone in cerebral smooth muscle COMMENTARY Christian Soellera,1 + Ryanodine receptors (RyRs) are large intracellular Ca2 WT mdx channels that provide the molecular basis of the pro- K+ K+ higher BK + + + cess termed Ca2 -induced Ca2 release (1). Ca2 sig- activity naling through RyRs has been shown to be critical for CaC BK BK skeletal, cardiac, and smooth muscle physiology (2) as well as for neurons (3) and secretory cells like pancre- larger RyR atic beta cells. RyRs were first seen in the electron RyRs RyRs clusters microscope as ∼30-nm-size particles (4), and it had SMCs been known from EM studies that they form clusters, SR SR typically in the narrow “junctional” space between sar- colemma and sarcoplasmic reticulum (SR) membranes. + Due to the inherent positive feedback of Ca2 -induced + Ca2 release, the size and shape of RyR clusters are expected to affect their functional activity. Indeed, de- SMCs SMCs tailed mathematical modeling suggests that RyR cluster- cerebral artery ing is important for determining the excitability of RyR- + mediated Ca2 release (5, 6) and could, in principle, also + affect the amount of Ca2 released in microscopic re- 2+ lease events termed Ca sparks (7). The experimental myogenic tone study of RyR clustering has received a boost with the reduced advent of optical superresolution microscopy, which myogenic has made the assessment of RyR cluster size more acces- sible by using essentially standard immunolabeling pro- tone tocols. When first applied in cardiac muscle cells, this myogenic tone (%) tone myogenic approach revealed a broad, approximately exponential vessel pressure (%) tone myogenic vessel pressure RyR cluster size distribution (8), which had previously not Fig.